The Complete Guide to 10x Genomics scATAC-seq: From Library Prep to Data Interpretation
This comprehensive guide details the 10x Genomics Chromium Single Cell ATAC solution for profiling chromatin accessibility at single-cell resolution.
The Complete Guide to 10x Genomics scATAC-seq: From Library Prep to Data Interpretation
Abstract
This comprehensive guide details the 10x Genomics Chromium Single Cell ATAC solution for profiling chromatin accessibility at single-cell resolution. Targeted at researchers and drug development professionals, it covers foundational principles of assay for transposase-accessible chromatin (ATAC) with sequencing, provides a step-by-step workflow from nuclei isolation to library preparation, addresses common troubleshooting and optimization strategies, and validates the technology's performance through data quality metrics and comparative analysis with other methods. The article synthesizes practical insights for experimental design, data interpretation, and translational applications in immunology, oncology, and neuroscience.
Understanding scATAC-seq: Deciphering Regulatory Genomics at Single-Cell Resolution
Within the context of a 10x Genomics single-cell ATAC-seq (scATAC-seq) workflow research thesis, understanding the core biochemical principle of the Assay for Transposase-Accessible Chromatin (ATAC-seq) is fundamental. This application note details the principle and protocols, focusing on how the method exploits hyperactive Tn5 transposase to tag and capture nucleosome-free, accessible genomic regions, which are pivotal for gene regulation studies in basic research and drug discovery.
Core Biochemical Principle
ATAC-seq identifies open chromatin regions by utilizing a genetically engineered, hyperactive Tn5 transposase. This enzyme is pre-loaded with sequencing adapters. It simultaneously cuts open, accessible DNA and inserts the adapters ("tagmentation") in a single enzymatic step. Regions densely packed with nucleosomes are inaccessible to Tn5, preventing tagmentation. The resulting fragments are then PCR-amplified and sequenced, providing a genome-wide map of chromatin accessibility.
Diagram 1: Tn5 tagmentation targets only accessible chromatin regions.
Detailed Protocol: Key Steps in scATAC-seq Library Prep
Nuclei Isolation and Tagmentation
Aim: Isolate intact nuclei and perform transposition. Reagents:
- Lysis Buffer (e.g., 10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, 0.1% IGEPAL CA-630, 1% BSA, 0.2U/µl RNase inhibitor).
- ATAC-seq Buffer (33mM Tris-acetate pH 7.8, 66mM Potassium acetate, 11mM Magnesium acetate, 16% DMF).
- Loaded Tn5 Transposase (commercially available or prepared in-house).
Procedure:
- Gently lyse cells in cold lysis buffer for 3-5 minutes on ice. Centrifuge to pellet nuclei.
- Resuspend nuclei pellet in ATAC-seq buffer mixed with loaded Tn5 transposase.
- Incubate at 37°C for 30-60 minutes with gentle mixing.
- Immediately purify DNA using a silica-membrane-based clean-up kit (e.g., MinElute PCR Purification Kit). Elute in low-EDTA TE buffer.
Post-Tagmentation Processing & 10x Barcoding
Aim: Amplify and barcode fragments for single-cell resolution. Procedure:
- Perform a limited-cycle PCR (e.g., 5 cycles) to add sequencing primer sites and amplify tagmented DNA.
- For scATAC-seq using the 10x Genomics Chromium Controller, combine barcoded gel beads, sample index PCR primers, and the PCR product from step 1 into a single master mix.
- Load the mix into a Chromium chip to generate single-cell GEMs (Gel Bead-in-Emulsions), where each fragment from a single cell receives a unique cellular barcode.
- Perform a larger-scale PCR (e.g., 12-14 cycles) to add sample indices and complete adapter sequences.
- Clean up libraries with SPRIselect beads and quantify via Bioanalyzer/Qubit.
Key Quantitative Metrics for Library QC: Table 1: Expected Library QC Metrics for scATAC-seq
| Metric | Ideal Range | Measurement Tool |
|---|---|---|
| Fragment Size Distribution | Strong peak ~200bp (nucleosome-free), periodicity at 200bp intervals | Agilent Bioanalyzer/TapeStation |
| Library Concentration | > 1.5 nM | Qubit dsDNA HS Assay / qPCR |
| Cell Complexity (Fraction of Reads in Peaks, FRiP) | > 15-20% for scATAC-seq | Sequencing data analysis |
| Estimated Number of Cells | Within 10% of target cell recovery | Sequencing data analysis |
Diagram 2: Simplified 10x scATAC-seq library preparation workflow.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for scATAC-seq
| Reagent | Function | Critical Note |
|---|---|---|
| Hyperactive Tn5 Transposase | Enzyme that cuts DNA and inserts sequencing adapters. The core of ATAC-seq. | Must be pre-loaded with oligonucleotide adapters for efficient tagmentation. |
| Nuclei Lysis Buffer (with detergent) | Gently lyses the cell membrane while keeping nuclear membrane intact. | Optimization of detergent concentration is crucial for specific cell types. |
| SPRIselect Beads | Size-selects DNA fragments and cleans up reactions. | Removes short fragments (<100bp) and enzymes, critical for fragment size distribution. |
| 10x Barcoded Gel Beads | Contains unique barcodes for single-cell partitioning within GEMs. | Essential for assigning reads to individual cells in the 10x platform. |
| Chromium Chip & Buffer Kit | Microfluidics system to partition cells into nanoliter-scale droplets (GEMs). | Enables high-throughput single-cell analysis. |
| Next-Generation Sequencing Kits | For Illumina platforms (e.g., NovaSeq, NextSeq). | Paired-end sequencing (e.g., 50 bp x 2) is standard for insert size analysis. |
Bulk assays, while foundational, average signals across thousands to millions of cells, obscuring critical heterogeneity inherent in biological systems—be it in tumor microenvironments, immune responses, or developmental lineages. This application note, framed within our thesis on advancing 10x Genomics scATAC-seq workflows, details how single-cell chromatin accessibility profiling transcends bulk ATAC-seq by enabling the discovery of rare cell states, delineating regulatory dynamics, and constructing predictive models of gene regulation at cellular resolution. This is critical for identifying novel drug targets and understanding mechanisms of resistance in therapeutic development.
Quantitative Comparison: Bulk ATAC-seq vs. scATAC-seq
The following table summarizes key performance and output metrics, illustrating the superior informational depth of single-cell approaches.
Table 1: Comparative Analysis of Bulk ATAC-seq and 10x Genomics scATAC-seq
| Metric | Bulk ATAC-seq | 10x Genomics scATAC-seq | Implication for Research |
|---|---|---|---|
| Cell Type Resolution | None (Averaged) | High (Thousands of cells individually profiled) | Identifies rare populations (<1% abundance) and continuous transitions. |
| Typical Peaks Called | 50,000 - 100,000 | 150,000 - 800,000 (aggregated from cells) | Uncovers a more comprehensive cis-regulatory landscape. |
| Key Output | A consensus accessibility profile | Cell-by-peak matrix, cell clustering, trajectory inference | Enables linking of regulatory variation to cellular phenotype. |
| Data Complexity | Low (One profile) | High (Multi-dimensional, sparse matrix) | Requires specialized bioinformatics pipelines (e.g., Cell Ranger, ArchR, Signac). |
| Typical Sequencing Depth | 20-50 million reads | 25,000-100,000 reads per cell (Total: 200M-1B reads) | Demands significant sequencing investment but yields multi-cell insights. |
| Primary Application | Defining shared regulatory elements | Mapping regulatory diversity, inferring gene regulatory networks (GRNs) | Directly identifies candidate regulators of cell fate decisions. |
Detailed Protocol: High-Viability Nuclei Isolation for scATAC-seq
Successful scATAC-seq hinges on high-quality, intact nuclei. This protocol is optimized for frozen tissue samples, a common scenario in translational research.
Application Note Protocol ANP-001: Nuclei Isolation from Frozen Tissue for 10x scATAC-seq
1. Principle: Gently lyse cellular membranes while keeping nuclear membranes intact, remove cytoplasmic debris, and resuspend nuclei in an isotonic buffer compatible with the 10x Genomics Chromium Next GEM technology.
2. Materials & Reagents:
- Fresh or snap-frozen tissue sample (≤ 25 mg)
- Homogenization Buffer: 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl₂, 0.1% IGEPAL CA-630, 1% BSA, 1 mM DTT, 0.1 U/µL RNase inhibitor, 1x protease inhibitor. Keep ice-cold.
- Wash Buffer: 1x PBS, 1% BSA, 0.1 U/µL RNase inhibitor. Keep ice-cold.
- Nuclei Buffer: 1x Diluted Nuclei Buffer (10x Genomics) or 1x PBS with 1% BSA.
- DAPI Solution (1 µg/mL)
- 40 µm Flow Cytometry Strainer
- Pre-chilled Dounce homogenizer (loose and tight pestles)
- Refrigerated centrifuge
3. Procedure: 1. Tissue Preparation: On dry ice, mince 10-25 mg of frozen tissue into small pieces in a petri dish. Transfer to a pre-chilled Dounce tube. 2. Homogenization: Add 1 mL of ice-cold Homogenization Buffer. Dounce with the loose pestle (10-15 strokes), then with the tight pestle (10-15 strokes). Monitor lysis visually. 3. Filtration: Filter the homogenate through a pre-wet 40 µm strainer into a new tube on ice. 4. Centrifugation: Spin at 500 rcf for 5 minutes at 4°C. Carefully aspirate supernatant. 5. Wash: Resuspend pellet in 1 mL ice-cold Wash Buffer by gentle pipetting. Centrifuge at 500 rcf for 5 minutes at 4°C. Aspirate supernatant. 6. Resuspension & Counting: Gently resuspend nuclei in 100-500 µL of Nuclei Buffer. Stain a 10 µL aliquot with DAPI and count using a hemocytometer or automated cell counter. Assess integrity via microscopy. 7. Quality Control: Target concentration: 1,000-10,000 nuclei/µL. Viability (intact nuclei) should be >90% by DAPI staining. Adjust concentration with Nuclei Buffer for 10x library preparation (target: 10,000 nuclei per reaction).
Visualization of Workflows and Pathways
Diagram 1: From Tissue to Regulatory Insights: scATAC-seq Workflow
Diagram 2: Decoding Chromatin Accessibility to Gene Regulation
The Scientist's Toolkit: Essential Reagents for scATAC-seq
Table 2: Key Research Reagent Solutions for Robust scATAC-seq
| Reagent / Material | Function & Role in Workflow | Critical for Overcoming |
|---|---|---|
| Chromium Next GEM Chip K | Microfluidic device to partition single nuclei into Gel Bead-In-Emulsions (GEMs) for barcoding. | Bulk processing; enables high-throughput, single-cell resolution. |
| Chromium scATAC-seq Library Kit | Contains all enzymes (Tn5 transposase) and buffers for tagmentation, barcode addition, and library PCR. | Inefficient and non-uniform tagmentation. |
| Nuclei Buffer & Lysis Reagents | Iso-osmotic buffers with detergents optimized for nuclear membrane integrity while removing cytoplasm. | Clogging of microfluidic chips; background from cytoplasmic debris. |
| High-Fidelity PCR Enzymes | Amplify low-input, tagmented DNA with minimal bias and duplicate reads. | Loss of library complexity and introduction of amplification artifacts. |
| SPRIselect Beads | Size-selective magnetic beads for post-reaction clean-up and size selection of final libraries. | Contaminant carryover and inappropriate fragment size distributions. |
| Dual Index Kit Sets | Provide unique sample indices for multiplexing multiple libraries in a single sequencing run. | Sample misidentification (index hopping) and reduced experimental throughput. |
| Cell Ranger ATAC Pipeline | Primary analysis software for demultiplexing, barcode processing, alignment, and peak calling. | Handling complex, sparse data formats and generating standardized output files for downstream analysis. |
Within the context of a thesis investigating the 10x Genomics single-cell ATAC-seq (scATAC-seq) workflow and library preparation, understanding the broader Chromium platform ecosystem is essential. This platform enables high-throughput, single-cell analysis of gene expression, immune profiling, chromatin accessibility, and more, revolutionizing research in oncology, immunology, neuroscience, and drug development.
The Chromium Platform Ecosystem: Core Components
Instrumentation & Controllers
- Chromium Controller: The core microfluidic instrument that partitions cells or nuclei into nanoliter-scale Gel Bead-In-EMulsions (GEMs). It is the physical platform for all single-cell assays.
- Chromium Connect: An automated system designed for hands-free, walk-away library preparation, reducing manual labor and increasing reproducibility.
Assay Portfolio
The platform supports a range of assays, each generating multiomic insights.
Table 1: Core 10x Genomics Chromium Assays (2024)
| Assay Name | Primary Output | Key Application in Research | Compatible with Fixed Cells? |
|---|---|---|---|
| Single Cell Gene Expression (3' & 5') | Transcriptome profiling | Cell type identification, differential expression, trajectory inference | Yes (with Fixation Kit) |
| Single Cell Immune Profiling | Paired V(D)J sequences + Gene Expression | T/B-cell clonality, antigen specificity, immune repertoire analysis | Yes |
| Single Cell ATAC-seq | Chromatin accessibility landscape | Regulatory element identification, cis-regulatory network inference | No (requires fresh/frozen nuclei) |
| Single Cell Multiome ATAC + Gene Exp. | Simultaneous chromatin accessibility + gene expression | Linked regulatory & transcriptional state in same cell | No |
| Visium Spatial Gene Expression | Whole transcriptome with spatial context | Tissue architecture analysis, spatially resolved cell typing | Yes (FFPE or Fresh Frozen) |
| Xenium In Situ | Subcellular spatial mapping of 100s of RNA targets | High-resolution spatial biology, cell neighborhood analysis | Yes (FFPE) |
Chemistry & Library Preparation
The platform utilizes a shared core chemistry: cells/nuclei are co-partitioned with uniquely barcoded Gel Beads and reagents. Within each GEM, reverse transcription or transposition occurs, tagging all nucleic acids from a single cell with a unique barcode. Post-partitioning, libraries are constructed for sequencing.
Software & Analysis
- Cell Ranger: Primary pipeline for processing raw sequencing data from Gene Expression, Immune Profiling, and ATAC-seq assays into feature-barcode matrices and V(D)J annotations.
- Loupe Browser: Interactive desktop software for visualization and analysis of 10x Genomics data.
- Space Ranger: Pipeline for processing Visium spatial data.
- Cloud Analysis: Web-based analysis tools (e.g., 10x Genomics Cloud) for accessible, scalable computing.
Detailed Protocol: 10x Genomics scATAC-seq Library Preparation
(Framed within a thesis on workflow optimization)
Principle: This protocol isolates nuclei, partitions them for transposition with barcoded Tn5 transposase, and prepares sequencing libraries to profile open chromatin regions at single-cell resolution.
I. Nuclei Isolation & Quality Control (Critical Step)
- Tissue Dissociation/Fresh Cell Preparation: Mechanically and enzymatically dissociate tissue to a single-cell suspension. For cell lines, harvest log-phase cells.
- Nuclei Extraction: Pellet cells (300-500 rcf, 5 min, 4°C). Lyse plasma membrane using chilled lysis buffer (e.g., 10mM Tris-HCl, 10mM NaCl, 3mM MgCl2, 0.1% IGEPAL CA-630, 1% BSA, 0.2U/µl RNase Inhibitor). Incubate on ice for 3-5 min.
- Nuclei Wash & Resuspension: Quench lysis with wash buffer (1% BSA in PBS). Pellet nuclei (500 rcf, 5 min, 4°C). Gently resuspend in Nuclei Buffer (1x PBS, 1% BSA, 0.2U/µl RNase Inhibitor).
- QC & Counting: Stain with Trypan Blue or DAPI. Count using a hemocytometer or automated cell counter. Assess integrity under a microscope. Target: ≥10,000 intact nuclei, >95% viability, minimal clumps.
- Adjust Concentration: Dilute nuclei to the target concentration in Nuclei Buffer (e.g., 1,000-10,000 nuclei/µl).
II. GEM Generation & Transposition on Chromium Controller
- Master Mix Preparation: On ice, combine in a tube:
Nuclei Suspension,10x Transposase Enzyme,ATAC Buffer. Mix gently. - Chip Loading: Load the master mix, Single Cell ATAC Gel Beads, and Partitioning Oil into a Chromium Single Cell ATAC Chip.
- Run on Controller: Place chip in the Chromium Controller and run the "Single Cell ATAC" program. This generates ~10,000 GEMs, where transposition occurs within each partition.
III. Post-GEM Cleanup & Amplification
- Break Emulsions: Transfer GEMs to a tube. Add Recovery Agent and mix. Use a magnet to separate beads. Discard supernatant.
- Post-Transposition Wash: Wash beads with Buffer EB.
- Library Amplification (PCR): Resuspend beads in Amplification Mix (PCR primers, PCR enzyme, buffers). Perform PCR:
72°C for 5 min; 98°C for 30s; then [98°C for 10s, 63°C for 30s, 72°C for 1 min] for 11-14 cycles. - SPRIselect Cleanup: Purify amplified DNA using SPRIselect beads at a 0.6x and 1.2x ratio to remove short fragments and primers.
IV. Library Construction & QC
- Dual Indexing PCR: Add sample-specific Dual Indexes via a second, shorter PCR (e.g., 8 cycles).
- Final Cleanup: Perform a 0.6x and 1.2x SPRIselect bead cleanup.
- Quality Control: Assess library using a Bioanalyzer/TapeStation (expect a broad smear from ~200-1200 bp). Quantify via qPCR (Kapa Library Quant Kit) for accurate sequencing loading.
- Sequencing: Pool libraries and sequence on an Illumina platform. Recommended: Paired-end sequencing (read1: 50+ cycles, read2: 50+ cycles, i7 index: 8 cycles, i5 index: 16 cycles).
Key Signaling & Workflow Visualizations
Title: scATAC-seq Library Prep Core Workflow
Title: Core Components of the 10x Genomics Ecosystem
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents for 10x Genomics scATAC-seq Workflow
| Item | Function & Importance in scATAC-seq Thesis Research |
|---|---|
| Chromium Single Cell ATAC Kit | Contains all core reagents (Gel Beads, enzymes, buffers, primers) for library prep from nuclei. Essential for standardization. |
| Chromium Single Cell ATAC Chip | Microfluidic device for generating GEMs on the Controller. Specific to the ATAC assay. |
| Partitioning Oil | Immiscible oil used to create stable nanoliter-scale droplets (GEMs) on the chip. |
| SPRIselect Beads | Solid-phase reversible immobilization beads for size selection and cleanup of DNA libraries. Critical for removing primer dimers. |
| Nuclei Buffer (1% BSA/PBS + RI) | Resuspension buffer to maintain nucleus integrity, prevent clumping, and inhibit RNA degradation. |
| RNase Inhibitor | Prevents RNA degradation during nuclei isolation, crucial for preserving potential coupled RNA signals in future multiome work. |
| Dual Index Kit Set A | Provides unique combinatorial indexes for multiplexing multiple samples in a single sequencing run. |
| Kapa Library Quantification Kit | qPCR-based kit for accurate molar quantification of final libraries, ensuring optimal sequencing cluster density. |
| High-Sensitivity DNA Assay (Bioanalyzer) | Electrophoresis-based QC to assess library fragment size distribution and detect adapter contamination. |
Application Notes
This document details the application of the 10x Genomics Single Cell ATAC-seq (scATAC-seq) workflow within a research thesis focused on optimizing library preparation and analytical pipelines for chromatin accessibility mapping. The primary objective is to enable high-resolution deconvolution of cellular heterogeneity, dynamic state transitions, and underlying gene regulatory networks, providing critical insights for target discovery and biomarker identification in drug development.
Core Quantitative Metrics: The following table summarizes key performance and output metrics from a standard 10x Genomics Chromium Next GEM scATAC-seq experiment, based on current manufacturer specifications and recent peer-reviewed studies.
Table 1: scATAC-seq Experiment Performance Metrics
| Metric | Typical Range/Value | Description/Impact |
|---|---|---|
| Cells Recovered | 5,000 - 15,000 per lane (Chromium X) | Defines experimental scale and statistical power for rare population detection. |
| Nuclei Passed QC | 60-80% of loaded | Indicates quality of nuclei isolation and staining protocol. |
| Median Fragments per Cell | 20,000 - 60,000 | Critical for assessing sequencing depth and signal-to-noise ratio per cell. |
| Fraction of Fragments in Peaks (FRiP) | 20-50% | Key QC metric for signal enrichment; >15% is generally acceptable. |
| Transcription Start Site (TSS) Enrichment Score | 8 - 20+ | Measures signal-to-background; higher scores indicate cleaner data. |
| Peak-Cell Matrix Sparsity | >99% | Inherent characteristic of scATAC data requiring specialized analytical tools. |
| Thesis Focus: Library Complexity | Non-Redundant Fraction (NRF) | A key thesis metric: NRF > 0.8 indicates high-complexity libraries, minimizing PCR duplicates. |
Key Applications:
- Cell Type & State Mapping: Unsupervised clustering based on integrated chromatin accessibility profiles enables identification of distinct cell types and subpopulations without prior marker knowledge. Pseudotime trajectory inference can map continuous state transitions (e.g., differentiation, activation).
- Regulatory Landscape Deconvolution: Cell-type-specific open chromatin regions are mapped to putative cis-regulatory elements (enhancers, promoters). Motif enrichment analysis within these regions identifies active transcription factors (TFs) driving cell identity.
- Integration with scRNA-seq: Paired or integrated analysis with gene expression data links regulatory elements to target genes, constructing cell-type-specific gene regulatory networks (GRNs).
Detailed Protocols
Protocol 1: Nuclei Isolation & Preparation for 10x scATAC-seq
This protocol is optimized for fresh/frozen mammalian tissues (e.g., mouse spleen) as per the thesis's comparative analysis of dissociation methods.
Essential Materials (Scientist's Toolkit):
- Research Reagent Solutions:
- Nuclei Isolation & Wash Buffer: 10mM Tris-HCl (pH 7.4), 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1% Nonidet P40 Substitute, 1% BSA, 1U/µL Protector RNase Inhibitor. Function: Maintains nuclear integrity while lysing cytoplasmic membranes.
- 1x Diluted Nuclei Buffer (10x Genomics PN-2000207): Contains reagents to stabilize nuclei for gel bead-in-emulsion (GEM) generation.
- Tagmented Nuclei Wash Buffer: 10mM Tris-HCl (pH 7.4), 10mM NaCl, 3mM MgCl2, 1% BSA. Function: Removes transposase (Tn5) post-tagmentation to halt reaction.
- DAPI Staining Solution (1 µg/mL): For viability assessment via flow cytometry or fluorescence microscopy.
- 40µm Flowmi Cell Strainer: To obtain a single-nuclei suspension.
Methodology:
- Tissue Dissociation: Mince 25-50 mg of tissue with a scalpel in 1 mL of cold Nuclei Isolation Buffer on a Petri dish placed on ice.
- Homogenization: Transfer suspension to a 2mL Dounce homogenizer. Perform 15-20 strokes with the "loose" pestle (A), then 15-20 strokes with the "tight" pestle (B), keeping the tube on ice.
- Filtration & Centrifugation: Filter homogenate through a 40µm strainer into a 5mL LoBind tube. Centrifuge at 500 rcf for 5 min at 4°C.
- Red Blood Cell Lysis (if needed): Resuspend pellet in 2 mL of RBC Lysis Buffer (optional step for hematopoietic tissues). Incubate for 2 min on ice, then add 2 mL of Wash Buffer.
- Wash & Count: Centrifuge at 500 rcf for 5 min at 4°C. Resuspend pellet in 1 mL of 1x Diluted Nuclei Buffer. Count nuclei using a hemocytometer with DAPI staining. Assess integrity (intact, spherical) by microscopy.
- Quality Control: Target concentration: 1,000-10,000 nuclei/µL. Aim for viability >80% (DAPI-negative). Adjust concentration to 1,000-2,000 nuclei/µL for loading onto the Chromium chip.
Protocol 2: Library Preparation & QC (Chromium Next GEM)
Following the 10x Genomics Chromium Next GEM Single Cell ATAC Reagent Kits v2 (User Guide CG000209).
Key Steps:
- GEM Generation & Barcoding: Load the prepared nuclei suspension, Master Mix, and Gel Beads onto a Chromium Next GEM Chip. The instrument partitions single nuclei into Gel Bead-In-Emulsions (GEMs). Within each GEM, transposition occurs, fragmenting accessible DNA and simultaneously adding a shared i7 adapter and a unique barcode sequence.
- Post GEM-RT Cleanup & Amplification: Break emulsions, pool GEMs, and purify barcoded DNA fragments with Silane magnetic beads. Perform a limited-cycle (12-14 cycles) PCR to amplify library fragments and add sample index (i5) and P7 sequences.
- Library Cleanup & QC: Perform a double-sided SPRIselect bead cleanup (0.6x and 1.2x ratios) to size-select fragments (~200-1200 bp). Assess library quality using an Agilent TapeStation 4200 (High Sensitivity D1000 assay).
- Sequencing: Pool libraries and sequence on an Illumina NovaSeq 6000 or equivalent. Use the following thesis-optimized sequencing configuration:
- Read 1: 50 cycles (barcode + genomic DNA)
- i7 Index: 8 cycles (sample index)
- i5 Index: 16 cycles (sample index)
- Read 2: 50 cycles (genomic DNA)
Visualizations
Within the broader thesis on optimizing the 10x Genomics scATAC-seq workflow for library preparation, meticulous upfront planning is the single most critical determinant of success. This document outlines the essential pre-requisites regarding sample types and experimental design, framed as Application Notes and Protocols. The integrity of single-cell chromatin accessibility data is fundamentally reliant on the quality of the input nuclei suspension and the statistical robustness of the experimental plan.
Sample Type Specifications and Quality Metrics
The 10x Genomics Chromium Next GEM Single Cell ATAC Solution is designed for intact, fluorescently stained nuclei derived from a variety of fresh or frozen sample types. Not all tissues or cell sources are equivalent, and their inherent properties dictate specific isolation protocols.
Table 1: Compatible Sample Types and Key Characteristics
| Sample Type | Recommended Starting Cell Viability | Recommended Cell Input for Nuclei Isolation | Critical Pre-Processing Notes | Key QC Metric Post-Isolation |
|---|---|---|---|---|
| Fresh Primary Tissue (e.g., mouse brain, spleen) | >90% | 0.5-1 million cells | Mechanical dissociation must be optimized to minimize debris. Protease inhibitors recommended. | Nuclei yield (>50%), intact nuclear membrane (DAPI staining), minimal cytoplasmic debris. |
| Cryopreserved Primary Cells/Nuclei | N/A (for pre-frozen nuclei) | 0.5-1 million nuclei | Flash-freeze nuclei in suitable cryoprotectant (e.g., glycerol-based buffer). Thaw slowly on ice. | Post-thaw integrity, clump-free suspension, % viable nuclei by dye exclusion. |
| Cell Lines (Adherent/Suspension) | >95% | 0.5-2 million cells | Gentle trypsinization for adherent lines. Ensure single-cell suspension before lysis. | Nearly 100% nuclei release, low RNA contamination (low RNase treatment recommended). |
| PBMCs or Blood-Derived Cells | >95% | 1-2 million cells | Density gradient centrifugation essential. Red blood cell lysis may be required. | High nuclei purity, absence of granulocyte nuclei (if undesired), low clumping. |
| Fresh/Frozen Tissue for Nuclei Isolation | N/A | 10-50 mg tissue | Direct nuclei isolation via Dounce homogenization is often preferred over cell dissociation. | Nuclear concentration, size distribution, absence of tissue aggregates. |
Universal QC Requirement: Regardless of source, the final nuclei suspension must be:
- Concentration: 700-1,200 nuclei/µL in 1x Nuclei Buffer.
- Viability/Integrity: >90% as assessed by fluorescent dye (e.g., DAPI, Propidium Iodide) exclusion or imaging.
- Morphology: Singlets, free of clumps and excessive cytoplasmic debris.
- Buffer Compatibility: Resuspended in ice-cold, validated nuclei buffer (e.g., 10x Genomics Nuclei Buffer) without divalent cations that promote clumping.
Experimental Planning: Power Analysis and Replication
A common flaw in single-cell genomics is underpowered design. For a thesis aiming to derive biologically and statistically significant conclusions, the following must be planned a priori.
Table 2: Experimental Design Considerations for Comparative scATAC-seq Studies
| Design Factor | Recommendation | Rationale |
|---|---|---|
| Biological Replicates | Minimum n=3 independent biological samples per condition. | Accounts for biological variability. Pooling samples is not a substitute for true replication. |
| Target Cells per Sample | 5,000 - 15,000 high-quality nuclei loaded per channel. | Balances cost with ability to capture rare cell types (down to ~0.5% frequency). |
| Control Samples | Include a well-characterized cell line (e.g., HEK293T, GM12878) in each batch. | Serves as a technical control for Tn5 activity, library prep, and sequencing. |
| Batch Effects | Confound experiments by processing samples from all conditions simultaneously. If impossible, use a staggered design and plan for batch correction in analysis. | Technical variation from different reagent lots or processing days can obscure biological signals. |
| Sequencing Depth | Aim for 25,000-50,000 raw read pairs per nucleus after filtering. | Provides sufficient coverage across accessible regions for peak calling and clustering. |
| Cell Doublet Rate | Estimate expected doublets using the 10x Genomics Doublet Calculator. Aim for <5% per run. | Influenced by loaded nuclei concentration. Doublets confound downstream analysis. |
Protocol: Standardized Nuclei Isolation from Fresh Murine Spleen
This protocol is cited as a foundational method within the thesis for generating high-quality nuclei from a complex lymphoid tissue.
Title: Isolation of Single Nuclei from Murine Spleen for scATAC-seq. Objective: To reproducibly generate a clean, concentrated, and viable suspension of single nuclei compatible with the 10x Genomics Chromium Next GEM Single Cell ATAC workflow.
Materials (Research Reagent Solutions):
- Homogenization Buffer: Ice-cold, containing 10mM Tris-HCl (pH 7.5), 10mM NaCl, 3mM MgCl2, 0.1% Nonidet P40 Substitute, 1% BSA, 1mM DTT, 1x Protease Inhibitors. Function: Lyzes plasma membrane while stabilizing nuclear envelope.
- Nuclei Wash Buffer: Ice-cold, containing 10mM Tris-HCl (pH 7.5), 10mM NaCl, 3mM MgCl2, 1% BSA, 1x Protease Inhibitors. Function: Removes detergent and cytoplasmic contaminants without pelleting nuclei too hard.
- Nuclei Buffer (1x): Ice-cold 10x Genomics Nuclei Buffer or equivalent (e.g., 1x PBS + 1% BSA). Function: Final resuspension buffer compatible with the Chromium chip.
- Fluorescent Nuclear Stain: DAPI (1 µg/mL) or Propidium Iodide (1 µg/mL). Function: Allows visual assessment of nuclear integrity and counting via hemocytometer or flow cytometer.
- 40µm Cell Strainer. Function: Removes large aggregates and tissue debris.
- Refrigerated Centrifuge. Function: Maintains nuclei integrity during pelleting steps.
- Dounce Homogenizer (loose pestle). Function: Provides controlled mechanical lysis.
Methodology:
- Dissection & Dissociation: Euthanize mouse following approved IACUC protocol. Rapidly harvest spleen into a dish with 2-3 mL of ice-cold Homogenization Buffer. Gently tease the tissue apart with forceps.
- Mechanical Lysis: Transfer the tissue slurry to a Dounce homogenizer on ice. Perform 15-20 strokes with the loose pestle. Avoid excessive force.
- Filtration: Filter the homogenate through a pre-wet 40µm cell strainer into a 50mL conical tube on ice. Rinse the homogenizer with 2-3 mL of Homogenization Buffer through the strainer.
- Nuclei Washing: Centrifuge the filtered lysate at 500 rcf for 5 minutes at 4°C. Gently decant the supernatant. Resuspend the soft pellet in 5 mL of ice-cold Nuclei Wash Buffer by pipetting slowly. Repeat centrifugation.
- Final Resuspension & QC: Decant supernatant. Gently resuspend the pellet in 1 mL of ice-cold 1x Nuclei Buffer. Keep on ice.
- Quality Control: Stain a 10 µL aliquot with 1 µg/mL DAPI. Count and assess nuclei integrity using a hemocytometer under a fluorescent microscope. Intact nuclei will show bright, round, and singular DAPI staining. Adjust concentration to 700-1,200 nuclei/µL with 1x Nuclei Buffer. Proceed immediately to the Chromium controller or flash-freeze.
Visualizations
Title: Nuclei Preparation Decision Workflow
Title: End-to-End scATAC-seq Workflow from Plan to Data
Step-by-Step Protocol: Executing the 10x Genomics scATAC-seq Workflow
This application note details Phase 1 of a comprehensive study on optimizing the 10x Genomics Single Cell ATAC-seq (scATAC-seq) workflow for chromatin accessibility profiling in heterogeneous tissues. The isolation of high-quality, intact nuclei is the critical first step, directly impacting library complexity, data quality, and the success of downstream drug target identification. This protocol focuses on sample preparation and nuclei isolation tailored for challenging and precious clinical samples.
Table 1: Comparison of Nuclei Isolation Yields from Various Tissue Types
| Tissue Type | Starting Mass (mg) | Median Nuclei Yield (x10^3) | Viability (Trypan Blue) (%) | Recommended Lysis Buffer Incubation Time (min) |
|---|---|---|---|---|
| Fresh Mouse Cortex | 10 | 450 ± 50 | 98 ± 1 | 5 |
| Fresh Human PBMCs | 1x10^6 cells | 850 ± 100 | 99 ± 0.5 | 3 (on ice) |
| Frozen Human Tumor (OCT) | 25 | 220 ± 80 | 85 ± 10 | 8-10 |
| Flash-Frozen Mouse Spleen | 15 | 300 ± 60 | 92 ± 5 | 6 |
| Cultured Cell Line (Adherent) | 5x10^5 cells | 480 ± 70 | 97 ± 2 | 4 |
Table 2: Impact of Nuclei Integrity on 10x scATAC-seq Metrics
| Nuclei Quality Metric | Target Value | Pass Q.C. TSO Concentration (pM) | Post-GEM-RT Fragments per Nucleus (Mean) | FRIP Score* |
|---|---|---|---|---|
| Viability (Nuclei) | >90% | 0.8 - 1.2 | 12,500 - 25,000 | >0.4 |
| Viability (Nuclei) | 70-90% | 1.5 - 2.5 | 5,000 - 12,000 | 0.2 - 0.4 |
| Clumping/ Debris | High | Highly Variable | < 5,000 | <0.2 |
*FRIP: Fraction of Reads in Peaks.
Detailed Experimental Protocols
Protocol 1: Nuclei Isolation from Fresh/Frozen Mammalian Tissues
Objective: To extract intact, single nuclei from solid tissues for scATAC-seq. Materials: Dounce homogenizer (loose & tight pestles), Nuclei EZ Lysis Buffer (Sigma NUC-101) or equivalent, 1x PBS + 0.04% BSA, 40µm cell strainer, Refrigerated centrifuge.
- Tissue Mincing: Rapidly dissect ~10-25 mg of tissue on ice. Mince finely with a scalpel in a small volume of cold 1x PBS + 0.04% BSA.
- Homogenization: Transfer tissue slurry to a Dounce homogenizer containing 2 mL of chilled Nuclei EZ Lysis Buffer. Perform 15-20 strokes with the loose pestle (A), then 10-15 strokes with the tight pestle (B). Keep on ice.
- Lysis: Incubate the homogenate on ice for the duration specified in Table 1, depending on tissue type. Invert tube gently every 2 minutes.
- Filtration & Washing: Filter the lysate through a pre-wet 40µm cell strainer into a 15 mL conical tube. Wash with 3 mL of Lysis Buffer through the strainer.
- Pellet Nuclei: Centrifuge at 500 rcf for 5 minutes at 4°C. Carefully decant supernatant.
- Wash & Resuspend: Gently resuspend the pellet in 5 mL of 1x PBS + 0.04% BSA. Centrifuge at 500 rcf for 5 min at 4°C. Decant supernatant. Resuspend the final nuclei pellet in 100-500 µL of 1x PBS + 0.04% BSA. Keep on ice.
- Quality Control: Count and assess viability using Trypan Blue or Acridine Orange/Propidium Iodide (AO/PI) on a hemocytometer or automated cell counter. Target viability >90%, minimal clumps.
Protocol 2: Nuclei Isolation from Cryopreserved or Fixed Cells
Objective: To recover nuclei from archived samples (e.g., frozen cell pellets, Cytopreserved samples). Materials: As in Protocol 1, plus RNase inhibitor.
- Thawing: Rapidly thaw frozen cell pellet in a 37°C water bath until just a small ice crystal remains. Immediately transfer to 10 mL of cold 1x PBS + 0.04% BSA.
- Wash: Centrifuge at 300 rcf for 5 min at 4°C. Aspirate supernatant.
- Lysis: Proceed with lysis as in Protocol 1, Step 2-3, but consider shorter incubation (3-4 min on ice). For samples fixed with low concentrations of formaldehyde (e.g., <0.1%), a longer lysis (10-15 min) with gentle agitation may be required.
- DNase Treatment (Optional, for reduced ambient RNA): Resuspend nuclei pellet in 100 µL of PBS + 0.1 U/µL RNase-free DNase I. Incubate at room temp for 5 min. Quench with 5 µL of 0.5M EDTA.
- Final Wash & QC: Wash twice with 5 mL of 1x PBS + 0.04% BSA + 1 U/µL RNase inhibitor. Resuspend and perform QC as in Protocol 1.
Protocol 3: Critical Nuclei QC and Loading for 10x Chromium Chip
Objective: To accurately quantify and load viable nuclei into the 10x Chromium controller.
- Debris Removal: Gently layer nuclei suspension over a 1 mL cushion of 1x PBS + 1% BSA in a 1.5 mL tube. Centrifuge at 200 rcf for 3 min at 4°C. Carefully aspirate the top layer, leaving the pelleted nuclei.
- Final Resuspension: Resuspend nuclei in a precise volume of 1x PBS + 0.04% BSA to achieve a target concentration of 700-1,200 nuclei/µL. The ideal concentration minimizes doublet rate while ensuring efficient partitioning.
- Verification: Re-count using AO/PI staining. Calculate loading volume for the 10x Chromium Chip (e.g., 10µL for a target of 10,000 nuclei).
- Chip Loading: Mix nuclei gently with 10x ATAC Buffer and Enzyme from the Chromium Next GEM Single Cell ATAC Kit (v2 or latest). Load mixture into the designated well of a Chromium Chip G. Proceed immediately to the Chromium Controller for GEM generation.
Visualizations
Title: scATAC-seq Nuclei Isolation Core Workflow
Title: Critical QC and 10x Chip Loading Pathway
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for scATAC-seq Nuclei Isolation
| Item | Function in Protocol | Key Consideration |
|---|---|---|
| Nuclei EZ Lysis Buffer (Sigma) or Homogenization Buffer (10x) | Lyse plasma membranes while leaving nuclear membrane intact. Contains detergents and stabilizers. | Commercial buffers offer reproducibility. Buffer composition (e.g., salt, detergent conc.) may need optimization for specific tissue types. |
| Dounce Homogenizer (Glass) | Mechanical disruption of tissue architecture to release nuclei. | Use correct pestle clearance (loose then tight). Pre-chill. Avoid excessive force to prevent nuclear damage. |
| 1x PBS + 0.04% Bovine Serum Albumin (BSA) | Washing and resuspension buffer. BSA reduces nuclei sticking to pipettes and tubes. | Must be nuclease-free. 0.04% is optimal; higher % can interfere with 10x chemistry. |
| 40µm Cell Strainer | Removes large cellular debris, tissue clumps, and fiber aggregates. | Always pre-wet with buffer to minimize nuclei loss. Use low-protein-binding strainers. |
| Acridine Orange (AO) / Propidium Iodide (PI) Stain | Fluorescent viability dye for nuclei. AO stains all nucleic acids (green), PI stains DNA in dead/damaged nuclei (red). | Superior to Trypan Blue for nuclei, as it specifically stains DNA/RNA. Use with fluorescence-capable counter. |
| RNase Inhibitor | Protects accessible chromatin from RNA contamination and degradation during processing. | Critical when processing samples with high RNase activity (e.g., pancreas, spleen). Add to final wash/resuspension buffers. |
| Chromium Next GEM Single Cell ATAC Kit (10x Genomics) | Provides all specialized reagents, enzymes, and buffers for library construction post-nuclei isolation. | Kit version must be confirmed. Contains Tn5 transposase, unique barcodes, and PCR reagents. Requires -80°C storage. |
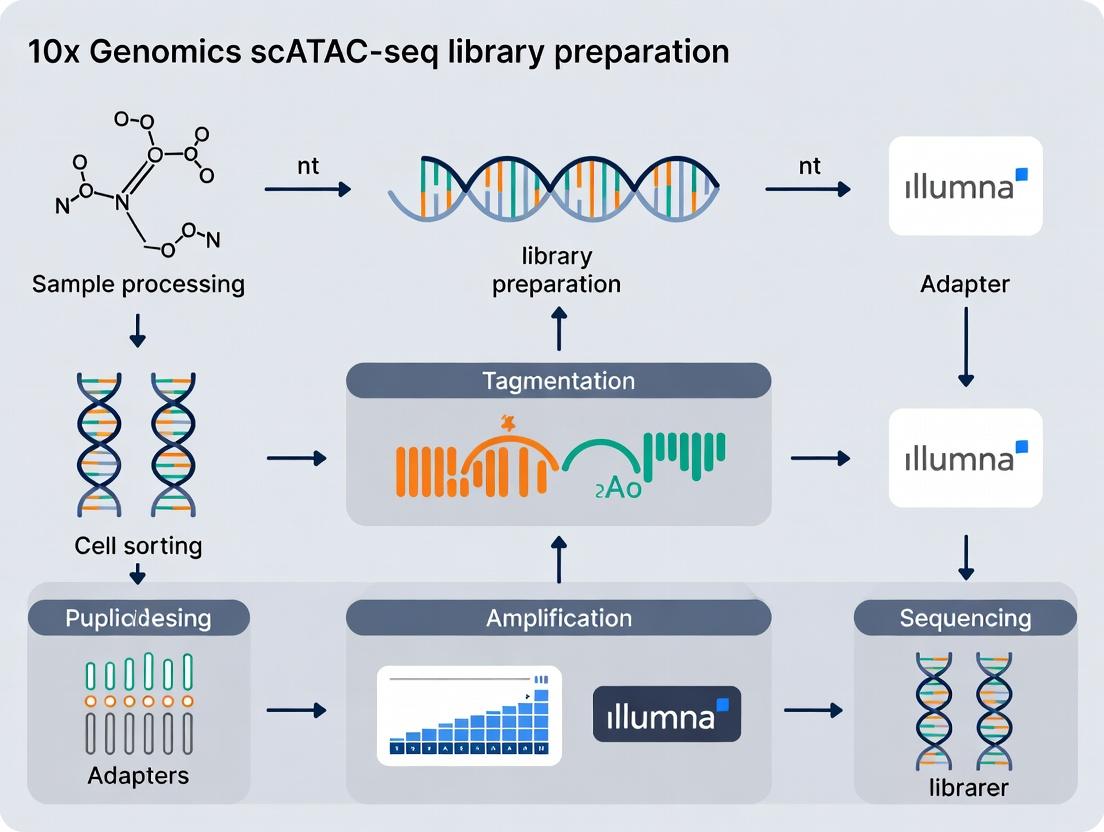
Within the comprehensive 10x Genomics Chromium Single Cell ATAC-seq workflow, Phase 2 represents the core enzymatic and compartmentalization step where fragmented, open chromatin is tagged with unique molecular identifiers. This phase directly follows nuclei isolation and permeabilization (Phase 1) and precedes PCR amplification and library sequencing (Phase 3). The transposition reaction, executed by a Tn5 transposase pre-loaded with mosaic ends, simultaneously fragments accessible genomic DNA and adds adapter sequences. These adapters are then linked to Gel Bead-derived barcodes and sample index sequences within uniquely formed GEMs, enabling the high-throughput, multiplexed analysis of chromatin accessibility landscapes from thousands of single cells.
Application Notes
The Transposition Reaction
The in situ tagmentation reaction is critical for targeting only nucleosome-free regions. The engineered Tn5 transposase exhibits optimal activity within the 10x Genomics reaction buffer, which maintains nuclear integrity while permitting enzyme access.
Key Quantitative Data for Transposition
Table 1: Optimized Transposition Reaction Parameters
| Parameter | Optimal Condition | Effect of Deviation |
|---|---|---|
| Incubation Temperature | 37°C | <37°C: Reduced efficiency; >37°C: Nuclear damage |
| Incubation Time | 60 minutes | <60 min: Incomplete tagmentation; >60 min: Increased background |
| Number of Nuclei | 500 - 10,000 per reaction | Lower: GEM recovery issues; Higher: Multiplets |
| Tn5 Enzyme Volume | As per kit (e.g., 2.5 µl) | Lower: Incomplete tagmentation; Higher: Increased di-tag artifacts |
| Reaction Buffer | Proprietary (Mg$^{2+}$ present) | Substitution leads to catastrophic failure |
GEM Generation and Barcoding
Following transposition and quenching, the reaction mixture is combined with Master Mix, Gel Beads, and partitioning oil on a Chromium chip. Each resulting GEM contains a single nucleus, a single Gel Bead, and reagents for barcoding.
Key Quantitative Data for GEM Generation
Table 2: GEM Generation and Barcoding Metrics
| Metric | Typical Yield/Value | Implication for Library Prep |
|---|---|---|
| Targeted GEM Recovery | ~100,000 GEMs per channel | Defines upper limit of cell recovery |
| Gel Bead Barcode Diversity | ~3.4 million unique barcodes | Ensves ultra-low barcode collision rate |
| Partitioning Efficiency | 1-10% cell capture per GEM | Minimizes multiplets (<0.9% per kit spec) |
| Barcoding Efficiency | >95% of fragments barcoded | Critical for high library complexity |
Detailed Experimental Protocols
Protocol: In-Nucleus Tagmentation Reaction
Objective: To fragment accessible DNA and add sequencing adapters. Materials: Permeabilized nuclei, Chromium Nuclei Buffer, Tn5 Transposase (from 10x kit), 0.2 mL PCR tubes, thermal cycler. Procedure:
- Prepare Master Mix: Combine on ice:
- 100 µL Nuclei Buffer
- 66 µL nuclease-free water
- 34 µL Tn5 Transposase Enzyme
- Total: 200 µL
- Combine with Nuclei: Transfer 20 µL of washed, permeabilized nuclei (500-10,000 nuclei) into a 0.2 mL PCR tube.
- Initiate Tagmentation: Add 20 µL of the prepared Master Mix to the nuclei. Pipette mix gently 10 times.
- Incubate: Place tube in a pre-warmed thermal cycler at 37°C for 60 minutes.
- Quench: Immediately add 40 µL of the provided Stop Solution. Mix by pipetting.
- Hold: Samples can be held at 4°C for up to 72 hours or proceed directly to GEM generation.
Protocol: GEM Generation and Barcoding via Chromium Controller
Objective: To partition single nuclei into droplets with uniquely barcoded Gel Beads. Materials: Tagmented nuclei reaction, Chromium Next GEM Chip K, 10x Gel Beads, Partitioning Oil, Recovery Reagents, Chromium Controller. Procedure:
- Prime Chromium Controller: Ensure the instrument is primed with partitioning oil according to the user guide.
- Prepare Chip: Load the Chromium Next GEM Chip K as follows:
- Well 1: 50 µL of Partitioning Oil.
- Well 2: 40 µL of the tagmented and quenched nuclei reaction.
- Well 3: 40 µL of Master Mix (from kit).
- Well 4: 10 µL of 10x Gel Beads.
- Well 5: 50 µL of Partitioning Oil.
- Run Chip: Place the loaded chip into the Chromium Controller and run the "Single Cell ATAC" program. This creates stable, barcoded GEMs.
- Recover Barcoded Fragments: Post-run, transfer the GEM emulsion (~100 µL) from the collection well to a new 1.5 mL tube.
- Break Emulsion: Add 125 µL of Recovery Agent, mix by inversion, and incubate at room temperature for 2 minutes.
- Purify DNA: Centrifuge at 16,000 rcf for 3 minutes. The aqueous layer (containing barcoded DNA) will be at the bottom (~130 µL). Carefully recover this layer, avoiding the oil and debris interphase.
- Cleanup: Proceed to SPRIselect bead cleanup as per the kit manual to purify barcoded fragments before amplification (Phase 3).
Visualizations
Diagram 1: scATAC-seq Phase 2 Workflow: Tagmentation to Barcoding.
Diagram 2: Oligo Structure and Barcoding Chemistry in a GEM.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Phase 2 Experiments
| Item | Function in Phase 2 | Critical Notes |
|---|---|---|
| 10x Chromium Next GEM Chip K | Microfluidic device to generate uniform GEMs. | Single-use; critical for partitioning efficiency. |
| 10x Gel Beads for scATAC-seq | Deliver barcodes, UMI, and primers. | Must be batch-controlled for consistent oligo loading. |
| Engineered Tn5 Transposase (10x) | Fragments accessible DNA and adds adapters. | Pre-loaded with mosaic ends; not interchangeable with homebrew Tn5. |
| Chromium Nuclei Buffer | Maintains nuclear integrity during tagmentation. | Proprietary formulation with optimized Mg$^{2+}$ concentration. |
| Partitioning Oil | Creates stable emulsion for GEM formation. | Viscosity and surfactant content are optimized for 10x chips. |
| Chromium Controller | Instrument for automated, reproducible GEM generation. | Requires regular priming and maintenance. |
| SPRIselect Beads | Post-GEM cleanup of barcoded fragments. | Size selection is crucial to remove primer dimers and excess oligos. |
| Stop Solution | Halts Tn5 activity post-tagmentation. | Contains SDS; critical for reproducibility. |
Within the broader 10x Genomics scATAC-seq workflow, Phase 3 is the final wet-bench stage where indexed DNA fragments from partitioned droplets are converted into sequencer-ready libraries. This phase involves the enzymatic construction of sequencing-compatible molecules, their amplification via PCR, and rigorous quality control to ensure library integrity, complexity, and optimal loading for next-generation sequencing. Success here directly determines data quality for downstream bioinformatic analysis in drug target discovery and regulatory genomics research.
Key Processes and Quantitative Benchmarks
Library Construction
Following droplet partitioning and barcoding (Phase 2), Gel Bead-in-EMulsions (GEMs) are broken, and the pooled post-ATAC material is purified. Library construction involves several enzymatic steps:
- Sample Index PCR: Addition of sample-specific dual index adapters (i5 and i7) via PCR. This enables multiplexing of multiple libraries on a single sequencing run.
- SPRIselect Clean-up: Size-selective purification using magnetic beads to remove primer dimers, very short fragments, and enzyme residues.
Table 1: Library Construction Reagent Functions
| Reagent/Solution | Function in scATAC-seq |
|---|---|
| Dual Index Kit (SI) | Contains unique i5 and i7 index primer pairs for sample multiplexing and sample tracking post-sequencing. |
| PCR Mix | Contains a high-fidelity DNA polymerase optimized for amplifying GC-rich, complex chromatin-derived libraries. |
| SPRIselect Beads | Perform size-selective cleanups to retain optimally sized library fragments (primarily ~200-1200 bp). |
| Buffer EB | Elution buffer (10 mM Tris-Cl, pH 8.5) for resuspending the final library post-purification. |
Library Amplification
The required number of PCR cycles is determined by the estimated cell count and must be optimized to prevent over-amplification (which increases duplicates) or under-amplification (which reduces complexity).
Table 2: Recommended PCR Cycles Based on Cell Recovery
| Estimated Number of Recovered Cells (Post-ATAC) | Recommended PCR Cycles for Library Amplification |
|---|---|
| ≤ 5,000 | 13 cycles |
| 5,001 – 10,000 | 12 cycles |
| 10,001 – 25,000 | 11 cycles |
| 25,001 – 50,000 | 10 cycles |
| > 50,000 | Consult 10x Genomics guidelines for cycle reduction. |
Quality Control (QC)
Comprehensive QC is non-negotiable prior to sequencing. Key metrics are assessed using bioanalyzer or tape station systems.
Table 3: Essential QC Metrics and Pass/Fail Criteria
| QC Metric | Method | Ideal/Primary Outcome | Acceptable Range |
|---|---|---|---|
| Library Concentration | Fluorometry (Qubit dsDNA HS Assay) | ≥ 1.5 ng/μL | 1.0 – 5.0 ng/μL |
| Fragment Size Distribution | Capillary Electrophoresis (Bioanalyzer High Sensitivity DNA) | Major peak ~200-600 bp; smear up to ~1200 bp. | Clear peak above primer dimer region (~145 bp). |
| Molarity for Sequencing | Calculation: (ng/μL × 10^6) / (average size bp × 650) | 5 – 50 nM for denaturation and loading. | Varies by sequencer. |
Detailed Experimental Protocol
Protocol: Chromatin Library Construction and Amplification (10x Genomics v2) This protocol follows GEM incubation and post-ATAC cleanup.
A. Sample Index PCR Setup
- Prepare Master Mix: On ice, combine the following in a nuclease-free PCR tube:
- Purified Post-ATAC DNA: X μL (entire yield from cleanup).
- Dual Index TT Set A: 2.5 μL.
- PCR Primer: 2.5 μL.
- PCR Mix: 25 μL.
- Nuclease-free Water: to a final volume of 50 μL.
- Thermal Cycler Program:
- 98°C for 45 seconds (initial denaturation).
- Cycle N times (see Table 2): 98°C for 20 seconds, 67°C for 30 seconds, 72°C for 1 minute.
- 72°C for 1 minute (final extension).
- Hold at 4°C.
B. Double-Sided SPRIselect Size Selection
- Right-Side (Large Fragment) Selection: Add 0.6X volume of resuspended SPRIselect beads to the 50 μL PCR reaction. Mix, incubate at RT for 5 minutes, pellet, and discard supernatant. This removes fragments < ~200 bp.
- Bead Wash: Wash beads on magnet with 80% ethanol twice. Air dry.
- Elute: Remove from magnet and elute DNA in 42.5 μL Buffer EB. Transfer supernatant to a new tube.
- Left-Side (Small Fragment) Selection: Add 0.15X volume of SPRIselect beads to the eluate. Mix, incubate, and pellet. SAVE THE SUPERNATANT. This step removes very large fragments and residual beads.
- Final Recovery: Transfer 40 μL of supernatant (containing size-selected library) to a new tube.
C. Library QC Analysis
- Quantification: Dilute 2 μL library 1:5 in Buffer EB. Use 2 μL of dilution for Qubit dsDNA HS assay per manufacturer's instructions.
- Size Profiling: Load 1 μL of undiluted library on a High Sensitivity DNA Bioanalyzer chip. Run and analyze profile.
- Molarity Calculation: Use concentration (ng/μL) and average size (bp) from bioanalyzer to calculate library molarity (nM). Adjust to loading concentration required by your sequencer.
Workflow and Pathway Visualizations
Phase 3 Library Construction & QC Workflow
Final scATAC-seq Library Structure
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Reagents for Phase 3
| Category | Item | Specific Function |
|---|---|---|
| Indexing & Amplification | Chromium i7 Multiplex Kit | Provides unique combinatorial dual indices for sample multiplexing. |
| Enzymatic Master Mix | KAPA HiFi HotStart ReadyMix | High-fidelity polymerase for minimal bias amplification of complex ATAC libraries. |
| Purification | SPRIselect Reagent Kit | Paramagnetic beads for reproducible, double-sided size selection. |
| Quantification | Qubit dsDNA HS Assay Kit | Fluorometric quantitation specific to dsDNA, insensitive to primers/adapter. |
| Size Analysis | Agilent High Sensitivity DNA Kit | Provides precise fragment size distribution and detects adapter dimer. |
| Buffers | Buffer EB (10 mM Tris-Cl, pH 8.5) | Low-EDTA elution buffer compatible with downstream enzymatic steps. |
Best Practices for Reagent Handling and Instrument Operation (Chromium Controller)
This document details essential protocols and practices for the 10x Genomics Chromium Controller within the context of single-cell ATAC-seq (scATAC-seq) workflow research. Proper reagent handling and instrument operation are critical for generating high-quality, reproducible chromatin accessibility data, which underpins downstream analyses in genomics research and drug discovery.
I. Reagent Handling: Core Principles & Protocols
Key Reagent Solutions & Materials
| Reagent/Material | Function | Critical Handling Notes |
|---|---|---|
| Chromium Next GEM Chip K | Microfluidic device for partitioning cells & reagents into Gel Bead-In-EMulsions (GEMs). | Store at 4°C. Equilibrate to room temp (RT) for 30 min before use. Inspect for bubbles/debris. |
| 10x Master Mix | Contains enzymes, co-factors, and polyacrylamide for GEM formation and ATAC reaction. | Always keep on a cold block (2-8°C). Vortex and spin down briefly before use. |
| Partitioning Oil | Immiscible oil phase for generating stable nanoliter-scale GEMs. | Store at 4°C. Protect from light. Vortex for 5 sec before loading. |
| Gel Beads (v2) | Barcoded beads with oligos for cell-specific labeling. | Store at -20°C in a non-frost-free freezer. Thaw on ice, then spin down. Keep cold until loaded. |
| Buffer & Enzyme Kits (ATAC) | For nuclei isolation, tagmentation, and library prep. | Aliquot enzymes to avoid freeze-thaw cycles. Store per manufacturer's spec. |
| Single Cell ATAC Library Kit | For post-GEM cleanup, amplification, and index PCR. | Store at -20°C. Thaw components on ice. Combine only immediately before use. |
| Dual Index Kit TT Set A | Provides unique dual indices for sample multiplexing. | Store at -20°C. Protect from light. Dilute as per protocol. |
| Reduced EDTA TE Buffer | For nuclei resuspension and dilution. | Ensure pH is 8.0. Filter through a 0.2 µm filter. |
Quantitative Stability Data
| Reagent | Storage Temp (°C) | Stable After Thawing (on ice) | Max Freeze-Thaw Cycles |
|---|---|---|---|
| 10x Master Mix | -20 to -80 | 2 hours | 2 |
| Gel Beads | -20 | 4 hours | Do not refreeze |
| Enzyme Mix (ATAC) | -20 | 1 hour | 1 |
| Library Amplification Mix | -20 | 2 hours | 3 |
| Partitioning Oil | 4 | 24 hours (RT) | N/A |
II. Chromium Controller Operation Protocol
Pre-Run Setup & Calibration
Objective: Ensure instrument is ready for a successful run. Materials: Chromium Controller, Computer with Controller Software, Chip K, Cables. Protocol:
- Power on the Chromium Controller and launch the software. Allow 10 minutes for system initialization.
- Perform a "Prime" operation using a new Chip K (without reagents) if the instrument has been idle >48 hours. This purges air from the fluidic lines.
- Visually inspect the instrument's seal for integrity. Clean with a dry, lint-free wipe if necessary.
- Confirm the instrument's internal temperature is stable at the set point (typically 21-28°C ambient).
Chip & Reagent Loading Protocol for scATAC-seq
Objective: Correctly load reagents to generate barcoded GEMs. Materials: Equilibrated Chip K, Master Mix (cold), Gel Beads (cold), Partitioning Oil, Prepared Nuclei Suspension (70-10,000 nuclei in 10-50 µL), Reagent Cooler. Protocol:
- Place the Chip K into the Reagent Cooler. Ensure it sits flat.
- Load Gel Beads: Pipette 40 µL of resuspended Gel Beads into the well marked "G". Avoid introducing bubbles.
- Load Master Mix: Pipette 27 µL of Master Mix into the well marked "M". Keep cooler lid closed between steps.
- Load Sample: Pipette 35 µL of prepared nuclei suspension into the well marked "S". Gently mix nuclei before loading.
- Load Partitioning Oil: Pipette 165 µL of Partitioning Oil into the well marked "O".
- Immediately place the loaded Reagent Cooler into the Chromium Controller. Close the lid.
- On the software, select the appropriate "Single Cell ATAC" protocol (e.g., "Chromium Next GEM Single Cell ATAC v2.0"). Click "Run".
Post-Run GEM Recovery & Cleanup Protocol
Objective: Harvest barcoded GEMs for downstream library preparation. Materials: Recovery Tubes (provided), 0.2 mL tube strip, Thermal Cycler, 10x Buffer CK, Silane Beads, SPRIselect Beads. Protocol:
- After the Controller run completes (≈12 min), carefully remove the Reagent Cooler.
- Using a P200 pipette set to 180 µL, gently aspirate the GEMs from the bottom of the well marked "▽". Avoid the oil layer.
- Transfer the GEMs to a 0.2 mL tube strip. Proceed immediately to the "GEM Incubation" step in the 10x ATAC protocol (37°C for 60 min in a thermal cycler with heated lid OFF).
- After incubation, add 50 µL of 10x Buffer CK and 125 µL of Recovery Agent to the GEMs. Mix by pipetting.
- Incubate at RT for 2 minutes. The oil/aqueous phase will separate.
- Transfer the ~300 µL of aqueous phase (bottom layer) to a new tube containing 200 µL of Silane Beads. Mix thoroughly.
- Follow the standard post-cleanup steps for PCR amplification and library construction as per the 10x Genomics User Guide (CG000492).
III. Workflow Visualization
Diagram Title: Core scATAC-seq Workflow with Chromium Controller
Diagram Title: GEM Formation & Barcoding Principle
Diagram Title: Pre-Run Reagent & Chip Quality Check
IV. Troubleshooting & Data Quality Metrics
Common Issues & Solutions
| Symptom | Potential Cause | Corrective Action |
|---|---|---|
| Low GEM Recovery Volume | Chip seal failure, air in lines. | Re-prime instrument. Ensure chip is fully seated in cooler. |
| High Oil in Recovery | Aspirated too close to oil layer. | Pipette more slowly; avoid top oil layer during recovery. |
| Low Nuclei Capture | Nuclei clumping, incorrect concentration. | Filter nuclei through a 40µm flowmi cell strainer. Re-count with trypan blue. |
| Failed Library | Enzyme degradation, poor GEM quality. | Ensure reagents are kept cold. Verify thermal cycler lid is OFF during GEM incubation. |
Expected Experimental Outcomes
| Metric | Target Range (scATAC-seq v2) | Measurement Method |
|---|---|---|
| Cells Recovered | 70-85% of loaded nuclei | Bioinformatics pipeline (Cell Ranger ATAC) |
| Median Fragments per Cell | 15,000 - 50,000 | Sequencing data analysis |
| Fraction of Fragments in Peaks (FRiP) | >15% | Sequencing data analysis |
| TSS Enrichment Score | >8 | Sequencing data analysis |
| Library Concentration | 2-10 nM | Qubit dsDNA HS Assay |
| Library Fragment Size | Major peak ~400-500 bp | Bioanalyzer/TapeStation HSD5000 |
Within the broader thesis on 10x Genomics scATAC-seq workflow optimization and library preparation research, this document provides critical Application Notes and Protocols for the downstream computational analysis and visualization steps. The transition from raw sequencing data to biologically interpretable insights is governed by the Cell Ranger ATAC pipeline and the Loupe Browser visualization suite. This analysis is foundational for research in chromatin accessibility, gene regulation, and cellular heterogeneity, directly informing target discovery and mechanistic studies in drug development.
Core Analytical Pipeline: Cell Ranger ATAC
Cell Ranger ATAC is a software suite that processes Chromium scATAC-seq data, aligning reads, calling peaks, counting fragments, and performing dimensionality reduction and clustering.
Detailed Protocol: Running Cell Ranger ATAC
Objective: To process raw FASTQ files from a 10x Genomics scATAC-seq experiment into a feature-barcode matrix and analysis results.
Materials & Computational Requirements:
- Input: Illumina sequencer-generated FASTQ files and a 10x-compatible sample index.
- Reference Genome: Pre-built 10x Genomics human (GRCh38) or mouse (mm10) reference package, or custom-built using
cellranger-atac mkref. - Software: Cell Ranger ATAC (latest version, e.g., 3.0.0).
- System: Linux-based high-performance computing environment with minimum 32GB RAM and 8 cores recommended.
Methodology:
- Data Organization: Place all FASTQ files in a single directory (
/path/to/fastqs/). Ensure files follow the naming convention:*_S1_L00[LANE]_[READ]_001.fastq.gz. - Pipeline Execution: Run the main pipeline command. Specify the sample ID, FASTQ path, and reference genome path.
- Output: The primary output is the
outsdirectory, containing:filtered_peak_bc_matrix.h5(Count matrix for downstream analysis).fragments.tsv.gz(Processed fragment file for genome browser viewing).clustering.csv&dimensionality_reduction.csv(Analysis results).web_summary.html(QC report).
Table 1: Key Metrics from Cell Ranger ATAC web_summary.html
| Metric | Description | Typical Target Range (Guideline) |
|---|---|---|
| Estimated Number of Cells | Cells identified by nuclei detection algorithm. | Protocol-dependent |
| Median Fragments per Cell | Sequencing depth per cell. | > 5,000 |
| Fraction of Fragments in Peaks | Proportion of reads falling in called peak regions. | > 0.15 - 0.30 |
| TSS Enrichment Score | Measure of signal-to-noise based on enrichment at transcription start sites. | > 6 - 10 |
| Total Fragments | Number of unique, valid fragment ends sequenced. | Experiment-dependent |
| FRIP (Fraction of Reads in Peaks) | Similar to "Fragments in Peaks," key QC metric. | > 0.15 - 0.30 |
Visualization and Exploration: Loupe Browser
Loupe Browser for scATAC-seq is a desktop graphical application for visualizing the results generated by Cell Ranger ATAC.
Detailed Protocol: Exploratory Analysis in Loupe Browser
Objective: To visually explore chromatin accessibility clusters, identify differentially accessible peaks, and link accessibility to gene annotation.
Methodology:
- Data Loading: Launch Loupe Browser. Use
File -> Opento select thecloupe.cloupefile from the Cell Ranger ATACoutsdirectory. - Cluster Exploration: In the "Cells" view, color cells by cluster (
k-meansorgraph-based) or by reduced dimensionality (t-SNE/UMAP). - Differential Accessibility Analysis: Select a cluster of interest. Right-click and choose "Find Differential Accessibility." The results table shows peaks significantly more accessible in the selected cluster versus all others (log-fold change, p-value, q-value).
- Genome Browser Integration: Double-click on a specific peak from the differential table. The integrated genome browser displays the accessibility track, fragment pileup, and nearby gene annotations.
- Gene Scoring & Annotation: Use the "Gene Accessibility" panel to view imputed gene activity scores. Correlate peak accessibility patterns with potential target gene expression.
The Scientist's Toolkit: Essential Research Reagent & Software Solutions
Table 2: Key Components for scATAC-seq Analysis Workflow
| Item | Function/Description |
|---|---|
| Chromium Next GEM Chip K | Part of library prep; partitions single nuclei with gel beads for barcoding. |
| Chromium Next GEM Reagent Kit | Contains enzymes and master mix for tagmentation and library construction. |
| Dual Index Kit TT Set A | Provides sample indexes for multiplexing libraries prior to sequencing. |
| Cell Ranger ATAC Software | Primary analysis pipeline for demultiplexing, alignment, peak calling, and count matrix generation. |
| Loupe Browser for scATAC-seq | Interactive visualization tool for exploring clusters, differential accessibility, and genomic regions. |
| 10x Genomics Reference Package | Species-specific genome reference with pre-computed indices for alignment and annotation. |
| High-Throughput Sequencer (e.g., Illumina NovaSeq) | Platform for generating paired-end sequencing reads from the final library. |
Workflow and Logical Diagrams
Diagram 1: From Sequencing to Insights: scATAC-seq Analysis Workflow
Diagram 2: Interpreting scATAC Data for Drug Discovery
Optimizing Your Experiment: scATAC-seq Troubleshooting and Peak Performance
Within the 10x Genomics scATAC-seq workflow, the quality of the initial nuclei suspension is the paramount determinant of final data quality. This thesis research identifies three interdependent pitfalls—low nuclei viability, suboptimal nuclei recovery, and elevated doublet rates—as critical failure points that compromise chromatin accessibility data, statistical power, and the accuracy of downstream biological interpretations in drug discovery and basic research.
Table 1: Impact of Common Pitfalls on scATAC-seq Outcomes
| Pitfall | Typical Metric Range (Problematic) | Target Metric Range (Optimal) | Primary Impact on Data |
|---|---|---|---|
| Nuclei Viability | <70% (DAPI+/PI+ nuclei) | ≥90% | High background reads, low library complexity, high mitochondrial signal. |
| Nuclei Recovery | <40% of input nuclei | 50-70% of input nuclei | Reduced cell number, lost rare populations, increased cost per cell. |
| Doublet Rate | >5% in loaded suspension | <1% in loaded suspension | Spurious trans-accessibility peaks, erroneous cell type calling. |
Table 2: Key Reagent Solutions for Mitigation
| Research Reagent / Material | Function in scATAC-seq Workflow |
|---|---|
| Nuclei Isolation & Storage | |
| Dounce Homogenizer (loose pestle) | Mechanically disrupts tissue without shearing nuclei. |
| Non-ionic Detergent (e.g., IGEPAL) | Gently lyses plasma membrane, leaving nuclear envelope intact. |
| Sucrose Gradient Solution | Purifies nuclei by density, removing cytoplasmic debris. |
| Nuclei Buffer (10x Genomics) | Stabilizes nuclei for short-term storage at 4°C. |
| Viability Assessment | |
| DAPI (4',6-diamidino-2-phenylindole) | Membrane-impermeant dye stains DNA of dead/damaged nuclei. |
| Propidium Iodide (PI) | Alternative membrane-impermeant DNA dye for viability staining. |
| Fluorescent Counting Beads | Enables absolute counting on a flow cytometer or automated counter. |
| Doublet Removal | |
| 40µm Flowmi Cell Strainer | Filters clumps prior to loading. |
| Fluorescence-Activated Cell Sorting (FACS) | Enables precise single-nucleus sorting based on DAPI intensity and pulse width. |
Detailed Application Notes & Protocols
Protocol A: High-Viability Nuclei Isolation from Murine Spleen
Objective: Isolate a suspension of single nuclei with ≥90% viability for 10x Chromium loading.
Materials: Cold PBS, Homogenization Buffer (10mM Tris-HCl pH7.5, 10mM NaCl, 3mM MgCl2, 0.1% IGEPAL, 1% BSA, 1U/µl RNase inhibitor), Wash Buffer (Homogenization Buffer without IGEPAL), 40µm strainer, Dounce homogenizer, centrifuge.
Method:
- Tissue Preparation: Minize 50mg fresh spleen in 1mL cold PBS on a petri dish. Transfer to a Dounce on ice.
- Homogenization: Add 1mL Homogenization Buffer. Dounce with loose pestle (A) 15-20 times. Do not over-homogenize.
- Filtration & Washing: Filter through a 40µm strainer into a 15mL tube. Add 10mL Wash Buffer.
- Centrifugation: Spin at 500 rcf for 5 min at 4°C. Carefully decant supernatant.
- Resuspension: Gently resuspend pellet in 1mL Wash Buffer with wide-bore pipette tips. Count and assess viability (Protocol B).
Protocol B: Accurate Nuclei Viability Assessment
Objective: Quantify the percentage of intact nuclei using membrane-impermeant dyes.
Materials: Nuclei suspension, DAPI (1µg/mL final concentration), Automated cell counter or flow cytometer.
Method:
- Staining: Combine 10µL nuclei suspension with 10µL DAPI solution. Incubate 3-5 min on ice.
- Counting: Load onto counting chamber or analyze by flow cytometry.
- Analysis: Viable nuclei are DAPI-negative (intact nuclear envelope). Dead/damaged nuclei are DAPI-positive. Calculate: % Viability = (DAPI-neg count / Total count) * 100.
Protocol C: Optimizing Nuclei Recovery for Loading
Objective: Maximize the number of viable nuclei loaded into the 10x Chromium chip.
Materials: Nuclei suspension, 0.04% BSA in PBS, Hemocytometer or automated counter, wide-bore tips.
Method:
- Accurate Concentration: Determine concentration using a hemocytometer. Account for viability. Target loading concentration is 7,000-10,000 nuclei/µL.
- Reduce Adhesion: Use buffers containing BSA (e.g., 0.04% BSA in PBS) for final resuspension and dilution.
- Pipetting Technique: Use wide-bore or low-retention tips for all handling steps post-isolation. Avoid vortexing; flick tube gently.
- Loading Calculation: Calculate volume needed for target recovery (e.g., 10,000 nuclei). Account for the Chromium's capture efficiency (~40-60%). Example: For a target of 5,000 recovered nuclei at 50% efficiency, load 10,000 nuclei.
Protocol D: Doublet Rate Minimization
Objective: Reduce the incidence of multiple nuclei encapsulated in a single droplet (GEM).
Materials: Pre-filtered nuclei suspension, 40µm Flowmi strainer, optional FACS sorter with 100µm nozzle.
Pre-Loading Filtration: Immediately before loading the Chromium chip, pass the concentrated nuclei suspension through a clean 40µm Flowmi strainer.
Optional FACS Sorting: Sort nuclei based on DAPI intensity and FSC-W vs. FSC-H to exclude doublets and debris. Collect into collection buffer with BSA.
Post-Cell Ranger Analysis: Utilize cellranger-atac aggr with the --normalize=none option and then apply tools like scrublet or ArchR's doublet scoring for in silico doublet identification and removal.
Visualization of Workflows and Relationships
Title: Interrelationship of scATAC-seq Sample Prep Pitfalls
Title: Optimal Nuclei Prep Workflow for 10x scATAC-seq
Diagnosing and Solving Library QC Issues (Fragment Size, Concentration)
Within a broader thesis on optimizing single-cell Assay for Transposase-Accessible Chromatin (scATAC-seq) workflows, library quality control (QC) stands as a critical gatekeeper. The 10x Genomics Chromium platform enables high-throughput profiling of chromatin accessibility; however, the success of downstream sequencing and analysis is wholly dependent on the quality of the input library. Two paramount QC metrics are library fragment size distribution and concentration. An incorrect fragment size (e.g., excess short fragments or sheared high molecular weight DNA) can lead to poor sequencing performance on the NovaSeq 6000 or NextSeq 2000 platforms, while inaccurate concentration measurement results in either wasted sequencing capacity or insufficient data yield. This application note details diagnostic procedures and solutions for these common issues, framed within scATAC-seq library preparation research.
Table 1: Acceptable vs. Problematic QC Ranges for 10x scATAC-seq Libraries
| QC Metric | Instrument/Method | Target Range (Ideal) | Sub-Optimal Range | Failure Range (Requires Remediation) |
|---|---|---|---|---|
| Fragment Size Profile | Agilent TapeStation / Bioanalyzer | Primary peak: 300 - 700 bp. Smear or ladder pattern expected. | Primary peak < 200 bp or > 1000 bp. | Single sharp peak < 150 bp (potential primer-dimer) or > 1500 bp. |
| Average Fragment Size (bp) | Agilent TapeStation / Bioanalyzer | 400 - 600 bp | 200 - 300 bp or 700 - 900 bp | < 180 bp or > 1000 bp |
| Molar Concentration (nM) | Qubit Fluorometer & qPCR (e.g., Kapa) | Qubit: 1-10 ng/µL. qPCR: 2-20 nM (for loading). | Qubit: 0.1-1 ng/µL. qPCR: 0.5-2 nM. | Qubit: < 0.1 ng/µL. qPCR: < 0.5 nM. |
| Concentration Discrepancy (Qubit vs qPCR) | Comparison of Assays | qPCR concentration ≤ 2x Qubit-derived molarity. | qPCR 2x - 4x Qubit-derived molarity. | qPCR > 4x Qubit-derived molarity (high adapter dimer). |
| DIN/DIQ Score | Agilent TapeStation | ≥ 7.5 for genomic DNA pre-fragmentation | 4.0 - 7.4 | ≤ 4.0 |
Table 2: Impact of Common QC Failures on 10x scATAC-seq Data
| QC Failure Symptom | Likely Cause | Downstream Sequencing Impact | Proposed Solution Pathway |
|---|---|---|---|
| Sharp peak ~150 bp | Excessive adapter-dimer or primer-dimer | Dominates sequencing reads, drastically reduces usable read depth. | A1, B3 |
| Broad smear < 300 bp | Over-fragmentation during tagmentation or sonication. | Low library complexity, poor peak calling in ATAC-seq data. | A2, B1 |
| High molecular weight (>1000 bp) | Incomplete tagmentation or fragmentation. | Low library yield, poor nucleus/cell recovery. | A2, B2 |
| High Qubit, very low qPCR | Significant contaminating dsDNA (e.g., gDNA, RNA) or degraded DNA. | Overestimation of library, severe under-clustering on flow cell. | A3, B4 |
| Low Qubit and low qPCR | Low cell input, poor tagmentation efficiency, or purification losses. | Low sequence yield, wasted flow cell lanes. | A4, B1, B4 |
Detailed Experimental Protocols
Protocol A: Diagnostic QC Procedures
A.1. Fragment Size Analysis via Agilent TapeStation 4200
- Purpose: Accurately determine library fragment size distribution and detect contaminants.
- Materials: Agilent TapeStation 4200, Genomic DNA ScreenTape & Reagents, library sample.
- Procedure:
- Thaw Genomic DNA ScreenTape, buffers, and ladder at room temperature for 30 min.
- Vortex ladder and buffer D, spin down.
- Prepare ladder: Add 15 µL buffer D to a strip tube, add 1 µL ladder, mix by pipetting.
- Prepare samples: For each library, add 15 µL buffer D to a strip tube, add 1 µL library, mix.
- Load tape into TapeStation, load plate, and run the "Genomic DNA" assay.
- Analysis: In the software, review the electrophoretogram and gel image. The expected scATAC-seq profile is a broad smear from ~200 bp to 1000+ bp. Note the average fragment size and the presence of any sharp low-molecular-weight peaks.
A.2. dsDNA Quantification via Qubit Fluorometer 4
- Purpose: Measure double-stranded DNA concentration, insensitive to free adapters/primers.
- Materials: Qubit 4 Fluorometer, Qubit dsDNA HS Assay Kit, library sample.
- Procedure:
- Prepare Qubit working solution by diluting dsDNA HS Reagent 1:200 in Buffer.
- Prepare standards: Add 190 µL working solution to each of two tubes, add 10 µL of standard #1 or #2.
- Prepare samples: Add 198 µL working solution to sample tubes, add 2 µL of undiluted library. Note: For low-yield libraries, use 5 µL of library + 195 µL solution.
- Vortex all tubes 2-3 sec, incubate 2 min at RT.
- Read on Qubit using the "dsDNA HS" assay. Calculate original concentration considering dilution factor.
A.3. Functional Library Quantification via qPCR (Kapa Library Quant Kit)
- Purpose: Quantify only library molecules competent for amplification and cluster generation.
- Materials: Kapa Library Quantification Kit for Illumina (ABI Prism), compatible qPCR instrument, library sample.
- Procedure:
- Perform a 1:10,000 serial dilution of the library in 10 mM Tris-HCl, pH 8.0.
- Prepare Kapa SYBR Fast qPCR Master Mix according to kit instructions.
- Prepare standards (provided) and diluted library samples in triplicate in a qPCR plate.
- Run qPCR with cycling conditions: 95°C for 5 min, then 35 cycles of (95°C for 30 sec, 60°C for 45 sec).
- Analysis: Generate a standard curve from the known standards. The qPCR software will output the concentration (in nM) of the diluted sample. Multiply by the dilution factor to obtain the original library concentration.
Protocol B: Remediation Protocols for Common Issues
B.1. Problem: Low Yield/Concentration (All Assays)
- Protocol: Re-Amplification of scATAC-seq Library
- Set up a 50 µL PCR reaction: 25 µL of 2x Kapa HiFi HotStart ReadyMix, 5 µL of 10 µM SI-PCR primer, 20 µL of purified library.
- Cycle: 98°C 45s; 6-8 cycles of (98°C 15s, 67°C 20s, 72°C 30s); 72°C 1 min.
- CRITICAL: Minimize cycles to avoid skewing representation. Perform a side-by-side qPCR on the pre- and post-amplification product to calculate the precise fold-increase.
- Purify using a 0.6x ratio of SPRIselect beads to remove excess primers and small fragments. Elute in 20 µL EB buffer.
B.2. Problem: High Molecular Weight Fragments/Incomplete Tagmentation
- Protocol: Optimization of Tagmentation Reaction Conditions
- Titrate Tn5 Enzyme: In a fresh experiment, prepare a series of tagmentation reactions using 20%, 50%, 100%, and 150% of the recommended Tn5 transposase volume.
- Optimize Incubation: Test tagmentation incubation times (5, 10, 30 min) at 37°C.
- Follow the standard 10x post-lysis and purification steps.
- Run QC on the resulting libraries to identify conditions yielding the ideal 300-700 bp peak.
B.3. Problem: High Adapter-Dimer Peak (~150 bp)
- Protocol: Double-Sided SPRIselect Size Selection
- Bring the library volume to 50 µL with nuclease-free water.
- Perform a right-side (upper cutoff) selection: Add 0.4x volumes of SPRIselect beads (20 µL to 50 µL sample). Mix, incubate 5 min, pellet, and SAVE THE SUPERNATANT containing the desired smaller fragments.
- Perform a left-side (lower cutoff) selection: To the supernatant, add 0.3x volumes of additional SPRIselect beads (relative to the original 50 µL volume: 0.3 x 50 µL = 15 µL). Mix, incubate, pellet.
- Discard this supernatant (which contains the adapter-dimers). Wash pellet twice with 80% ethanol.
- Elute in 25 µL EB buffer. Re-analyze on TapeStation.
B.4. Problem: Large Discrepancy Between Qubit and qPCR Values
- Protocol: Purification and Concentration of Libraries via Ethanol Precipitation
- To the library, add 0.1 volumes of 3M sodium acetate (pH 5.2) and 2.5 volumes of 100% ice-cold ethanol.
- Mix well and incubate at -20°C for 30+ minutes.
- Centrifuge at >12,000 g for 15 min at 4°C. Carefully remove supernatant.
- Wash pellet with 500 µL of 80% ice-cold ethanol. Centrifuge 5 min. Carefully remove all ethanol.
- Air-dry pellet for 5-10 min. Resuspend in 10-20 µL of 10 mM Tris-HCl, pH 8.0.
- Re-quantify using both Qubit and qPCR. The ratio should normalize if contaminants were removed.
Mandatory Visualization
Diagram Title: Library QC Decision and Remediation Workflow
Diagram Title: Root Cause Analysis of Common Library QC Failures
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for scATAC-seq Library QC and Remediation
| Item | Supplier/Example | Primary Function in QC/Remediation |
|---|---|---|
| Agilent Genomic DNA ScreenTape | Agilent Technologies (5067-5365) | Provides high-resolution fragment size analysis for libraries, critical for diagnosing size issues. |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific (Q32851) | Accurate, dye-based quantification of dsDNA library concentration, unaffected by free adapters. |
| Kapa Library Quantification Kit | Roche (KK4824) | qPCR-based assay quantifying amplifiable library molecules; the gold standard for loading Illumina sequencers. |
| SPRIselect Beads | Beckman Coulter (B23318) | Paramagnetic beads for precise size selection (e.g., adapter-dimer removal) and purification. |
| Kapa HiFi HotStart ReadyMix | Roche (KK2602) | High-fidelity polymerase for minimal-cycle re-amplification of low-yield libraries without bias. |
| 10x Genomics SI-PCR Primer | 10x Genomics (from kit) | Primer for the final library amplification step; used in re-amplification protocols. |
| Low TE Buffer or EB Buffer | Thermo Fisher Scientific (12090015) | Elution buffer for purified DNA, maintaining stability and compatibility with downstream steps. |
| Nuclease-free Water | Various (AM9937) | Diluent for libraries and reagent preparation, ensuring no enzymatic degradation. |
Optimizing Transposition Time and Cell Input Numbers
Application Notes
Within the 10x Genomics single-cell ATAC-seq (scATAC-seq) workflow, the transposition reaction is a critical determinant of final data quality. This reaction, facilitated by the Tn5 transposase, simultaneously fragments chromatin and adds sequencing adapters to accessible DNA regions. Optimization of two key parameters—transposition time and cell input number—directly impacts library complexity, signal-to-noise ratio, and the cost-efficiency of experiments.
Table 1: Impact of Transposition Time on scATAC-seq Metrics (10,000 Cell Input)
| Transposition Time (minutes) | Median Fragments per Cell | Fraction of Fragments in Peaks (FIP) | TSS Enrichment Score | Notes |
|---|---|---|---|---|
| 30 | 12,500 | 0.28 | 8.2 | Lower complexity, potential under-saturation. |
| 60 (Standard) | 25,000 | 0.42 | 14.5 | Balanced yield and specificity. |
| 90 | 28,000 | 0.41 | 14.0 | Marginal increase in fragments; possible over-transposition background. |
| 120 | 26,500 | 0.38 | 13.2 | Increased background fragments, reduced specificity. |
Table 2: Effect of Cell Input Number on Data Recovery (60-min Transposition)
| Target Cell Input | Estimated Recovery Rate | Recommended Sequencing Depth per Cell | Key Consideration |
|---|---|---|---|
| 5,000 - 10,000 | 65-75% | 25,000 reads | Optimal for focused studies; high data quality per cell. |
| 20,000 - 30,000 | 50-60% | 20,000 reads | Cost-effective for population-scale studies; increased multiplets risk. |
| 50,000+ | 40-50% | 15,000-20,000 reads | Maximizes cell discovery; requires rigorous demultiplexing and doublet removal. |
Experimental Protocols
Protocol 1: Titration of Transposition Time
Objective: To determine the optimal duration of the Tn5 transposase reaction for a given cell type. Materials: Nuclei from fresh/frozen cells, 10x Genomics Chromium Next GEM Chip K, 10x ATAC Buffer Reagents, Tn5 Transposase (provided), PBS + 0.04% BSA. Procedure:
- Prepare nuclei suspension according to the 10x Genomics Demonstrated Protocol (CG000169). Quantify using a hemocytometer and dilute to 1,000 nuclei/µL in PBS + 0.04% BSA.
- For each time point (30, 60, 90, 120 min), aliquot nuclei for 10,000 target cell recovery.
- Combine nuclei with Transposition Mix (Tn5, Buffer) in a 96-well plate. Mix thoroughly by pipetting.
- Incubate plate on a thermal cycler at 37°C. Remove each aliquot at its designated time point.
- Immediately add provided Silane Magnetic Beads to stop the reaction. Place on a magnetic stand and proceed with washing as per the standard protocol.
- Continue with barcoding, amplification, and library construction steps identically for all samples.
- Sequence libraries and analyze using Cell Ranger ATAC. Compare Median Fragments per Cell and Fraction of Fragments in Peaks (FIP).
Protocol 2: Optimization of Cell Input Numbers
Objective: To assess the relationship between loaded cell number and final recovered cell yield/data quality. Materials: As above, with a consistent transposition time (e.g., 60 min). Procedure:
- Prepare a single, high-quality nuclei suspension. Confirm viability and absence of clumps.
- Using accurate dilution, create aliquots targeting 5,000, 10,000, 25,000, and 50,000 input nuclei.
- Process each input aliquot separately through the entire 10x scATAC-seq workflow (transposition, barcoding, cleanup, amplification).
- Perform dual-indexed library preparation and quantify by qPCR.
- Pool libraries equimolarly and sequence on a NovaSeq 6000 (or equivalent). Aim for a minimum of 20,000 raw read pairs per targeted cell.
- Process data through Cell Ranger ATAC (
cellranger-atac count). The Estimated Number of Cells reported is the recovery. Calculate recovery rate: (Estimated Cells / Target Input) * 100. - For each condition, evaluate the cell-containing droplet saturation curve and the multiplet rate (estimated by the
cellranger-atac aggrsubcommand or tools likeArchR/Signac).
Visualizations
Diagram 1: scATAC-seq Transposition Optimization Logic
Diagram 2: 10x scATAC-seq Core Workflow Steps
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for scATAC-seq Transposition Optimization
| Item | Function & Role in Optimization |
|---|---|
| 10x Genomics Chromium Next GEM Chip K | Generates single-cell, barcoded Gel Bead-in-emulsions (GEMs). Cell input number is determined here. |
| Tn5 Transposase (10x Custom) | Engineered enzyme for simultaneous fragmentation and adapter tagging. Activity duration is the key variable for time optimization. |
| Chromium Nuclei Buffer | Maintains nuclear integrity during suspension preparation, critical for accurate cell counting and input. |
| SPRIselect Magnetic Beads | Size-selects DNA fragments post-transposition. Cleanup efficiency affects background noise in final libraries. |
| ATAC PCR Primer Cocktail | Contains unique i5 and i7 sample index primers for post-barcoding amplification and library indexing. |
| Bioanalyzer/TapeStation | Essential for QC of final library fragment size distribution (typical peak ~300-600 bp). |
| Cell Ranger ATAC Software | Primary analysis pipeline for mapping, peak calling, and generating key QC metrics (FIP, TSS score). |
Mitigating Ambient RNA and Contamination Artifacts
In the 10x Genomics scATAC-seq workflow, ambient RNA and contamination artifacts pose significant threats to data integrity. Ambient RNA, originating from lysed cells, can be captured in droplet emulsion, leading to false gene expression signals. Contamination from genomic DNA, nucleases, or carryover between samples can compromise chromatin accessibility profiles and library quality. This document details protocols and application notes for mitigating these artifacts within the broader thesis research on optimizing single-cell epigenomic library preparation.
Quantification of Artifact Impact
The following table summarizes key quantitative data on artifact sources and their measured impact on scATAC-seq data quality.
Table 1: Impact of Common Artifacts on scATAC-seq Metrics
| Artifact Source | Typical Concentration/Level | Observed Effect on Data | Mitigation Stage |
|---|---|---|---|
| Ambient DNA/Chromatin | 0.1-0.5 ng/µL in supernatant | Up to 15% reduction in fraction of fragments in peaks | Cell Wash & Nuclei Isolation |
| DNase Contamination | Trace amounts | Complete degradation of accessible chromatin fragments | Reagent QC & Clean-up |
| Barcoding Bead Carryover | 0.01% carryover rate | ~0.5% doublet/multiplet rate increase | Post-GEM Cleanup |
| PCR Reagent Contaminants | Inhibitors in polymerase mix | 30-50% reduction in library yield | Library Amplification |
| Nuclei Multiplexing Index Hopping | 0.1-2% error rate | Misassignment of 1-200 cells per 10k | Sample Indexing PCR |
Detailed Experimental Protocols
Protocol 1: Nuclei Isolation and Wash for Ambient DNA Reduction
This protocol minimizes ambient chromatin from damaged cells prior to GEM generation.
Materials:
- Fresh or frozen tissue/cells
- Nuclei Isolation Kit (e.g., 10x Genomics Nuclei Isolation Kit)
- DPBS, nuclease-free
- 1% BSA in DPBS, nuclease-free
- 40 µm cell strainer
- Refrigerated centrifuge
Method:
- Prepare tissue or cell suspension according to standard dissociation protocols. Keep samples on ice.
- Lyse cells using the recommended lysis buffer for 3-5 minutes on ice. Monitor under a microscope for intact nuclei release.
- Quench the lysis reaction with 1% BSA in DPBS (1:5 v/v ratio).
- Filter the nuclei suspension through a pre-wet 40 µm cell strainer.
- Centrifuge the filtered nuclei at 500 rcf for 5 minutes at 4°C. Carefully aspirate the supernatant, leaving ~50 µL to avoid pellet disturbance.
- Critical Wash Step: Resuspend the nuclei pellet in 1 mL of 1% BSA/DPBS. Centrifuge again at 500 rcf for 5 minutes at 4°C. Aspirate supernatant completely.
- Repeat Step 6 for a total of two washes.
- Resuspend the final, clean nuclei pellet in a small volume of nuclei buffer. Count using a hemocytometer with trypan blue or an automated cell counter.
- Adjust concentration to the target input for the 10x Chromium controller (e.g., 1,000-10,000 nuclei/µL).
Protocol 2: DNase Decontamination of Reagents and Work Surfaces
Eliminates trace DNase activity that can degrade accessible chromatin fragments.
Materials:
- RNase Away or DNAZap solution
- Nuclease-free water
- UV crosslinker (optional)
- Dedicated, filtered pipette sets
Method:
- Surface Decontamination: Wipe down all work surfaces, pipettes, and tube racks with a commercial DNase/RNase decontamination solution. Allow to air dry.
- Reagent Preparation: Prepare all buffers and solutions using nuclease-free water and molecular biology-grade reagents in a pre-cleaned hood.
- Critical Tool Treatment: For non-disposable plasticware (e.g., tube holders), soak in a 0.1% Diethyl pyrocarbonate (DEPC)-treated water solution for 1 hour, then rinse thoroughly with nuclease-free water and autoclave.
- UV Treatment (Optional but Recommended): Place non-enzymatic, heat-stable reagents (e.g., salts, buffers) in open tubes in a UV crosslinker and expose to 254 nm light for 10 minutes to crosslink any contaminating nucleic acids.
Protocol 3: Post-GEM Cleanup and Doublet Mitigation
Reduces contamination from barcoding bead carryover and ambient material post-encapsulation.
Materials:
- Recovery Agent (from 10x kit)
- Silane magnetic beads
- DynaMag magnet
- Fresh 80% ethanol
- Elution buffer (10 mM Tris-HCl, pH 8.0)
Method:
- After GEM-RT incubation and cleanup according to the standard 10x protocol, proceed to the post-cDNA amplification step.
- Following amplification, add a 0.6x volume of silane magnetic beads to the purified cDNA. Mix thoroughly and incubate at room temperature for 5 minutes.
- Place the tube on a magnetic rack. After the solution clears, carefully remove and discard the supernatant.
- Critical Wash: With the tube on the magnet, add 150 µL of 80% ethanol without disturbing the bead pellet. Incubate for 30 seconds, then remove the ethanol. Repeat for a total of two ethanol washes.
- Air-dry the bead pellet for 2-3 minutes. Do not over-dry.
- Remove from the magnet and elute cDNA in the recommended elution buffer (e.g., 40 µL). Mix well and incubate for 2 minutes.
- Return the tube to the magnet. Transfer the purified supernatant containing cDNA to a new, nuclease-free tube. Proceed to library construction.
Visualization of Workflows and Relationships
Title: Artifact Sources, Mitigation Protocols, and Outcomes
Title: scATAC-seq Workflow with Critical QC Steps
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Artifact Mitigation in scATAC-seq
| Item | Function in Mitigation | Example Product/Type |
|---|---|---|
| Nuclease-free BSA (1% in PBS) | Blocks non-specific binding during nuclei wash; stabilizes nuclei. | Molecular Biology Grade BSA |
| DNase/RNase Decontamination Spray | Eliminates nuclease contamination from work surfaces and equipment. | RNase Away, DNAZap |
| Silane Magnetic Beads | Selective cleanup of post-GEM reactions; removes primers, salts, and contaminants. | SPRIselect, AMPure XP |
| Nuclei Isolation Buffer | Maintains nuclear integrity while lysing cytoplasmic membrane, minimizing ambient release. | 10x Nuclei Isolation Kit, Homemade Sucrose Buffer |
| Dual-Size Filter Strainers | Removes cell clumps (40 µm) and debris (20 µm) to prevent droplet clogging. | Pluristrainer, Flowmi cell strainers |
| Indexed PCR Adapters with Unique Dual Indexes (UDIs) | Drastically reduces sample index hopping and cross-contamination during pooling. | 10x Dual Index Kit, IDT for Illumina UDIs |
| Fluorescent Nuclear Viability Stain | Allows accurate counting of intact nuclei and assessment of preparation quality. | DAPI, NucBlue (Hoechst 33342) |
| High-Sensitivity DNA Assay Kits | Quantifies low-input pre-library and final library concentrations accurately. | Qubit dsDNA HS, Agilent High Sensitivity DNA Kit |
Advanced Tips for Challenging Samples (Tissues, Frozen Cells)
Within a broader thesis on optimizing the 10x Genomics scATAC-seq workflow for diverse biological specimens, this application note addresses the unique challenges posed by challenging samples like primary tissues and frozen cell stocks. Successful library preparation from these samples requires modifications to standard protocols to ensure high nuclei yield, integrity, and data quality.
Table 1: Common Challenges and Impact on scATAC-seq Data Quality
| Challenge | Typical Sample Source | Primary Effect | Measurable Impact (if unmitigated) |
|---|---|---|---|
| High RNase Activity | Pancreatic tissue | RNA degradation | Reduced nucleus integrity; <10% viable nuclei |
| Extracellular Matrix Density | Heart, tumor tissue | Low nuclei yield | Yield < 1000 nuclei/mg tissue |
| Cryopreservation Artifacts | Frozen PBMCs, cell lines | Nuclear clumping, membrane damage | Aggregates in data; >30% doublet rate |
| High Lipid Content | Brain tissue (white matter), adipose | Viscous lysate, nucleus trapping | 50-70% loss during filtration |
| Endogenous Nuclease Activity | Spleen, liver | DNA cleavage, fragment loss | Low sequencing library complexity |
Detailed Protocols
Protocol 1: Nuclei Isolation from Fibrous/Frozen Tissue
This protocol is optimized for muscle, heart, or frozen tissue pieces.
- Pre-chill Tools: Cool homogenizer (e.g., Dounce), tubes, and buffers on ice.
- Mechanical Disruption:
- For fresh tissue: Mince 1-2 mm³ piece in chilled PBS on a petri dish.
- For frozen tissue: Place ~25 mg chunk in chilled Petri dish. Add 1 mL Nuclei EZ Lysis Buffer (Sigma, NUC101) directly onto frozen tissue. Mince immediately as it thaws.
- Homogenize: Transfer tissue + buffer to Dounce. Use 15-20 strokes with loose pestle (A), then 10-15 with tight pestle (B).
- Incubate & Filter: Incubate lysate on ice for 5 min. Filter through a 40 μm Flowmi cell strainer into a new tube.
- Wash & Purify: Underlay filtrate with 1 mL of 1x PBS with 1% BSA and 0.2 U/μl RNase inhibitor. Centrifuge at 500 rcf for 5 min at 4°C. Carefully aspirate supernatant.
- Density Purification (Optional for debris): Resuspend pellet in 1 mL 0.1% BSA in PBS. Layer onto 1 mL of 29% iodixanol solution. Centrifuge at 10,000 rcf for 20 min at 4°C (brake off). Collect nuclei at interface.
- Resuspend & Count: Resuspend final pellet in 1x Nuclei Buffer (10x Genomics). Count with AO/PI dye on hemocytometer. Target concentration: 700-1200 nuclei/μL.
Protocol 2: Reviving Frozen Cells for scATAC-seq
For cryopreserved cell suspensions (e.g., PBMCs, cell lines).
- Rapid Thaw: Thaw vial in 37°C water bath until just a small ice crystal remains.
- Gentle Dilution: Slowly add 5 mL pre-warmed (37°C) recovery medium (RPMI + 20% FBS + 10 U/μl DNase I) dropwise to the vial.
- Centrifuge & Wash: Transfer to 15 mL tube. Add 5 mL more medium. Centrifuge at 300 rcf for 5 min.
- DNase Treatment: Aspirate supernatant. Resuspend pellet in 1 mL of pre-warmed recovery medium. Incubate at room temperature for 10 min to dissociate aggregates.
- Wash & Lyse: Add 10 mL cold PBS + 0.04% BSA. Centrifuge at 500 rcf for 5 min. Aspirate. Perform nuclei isolation using standard 10x Genomics lysis buffer (10 min on ice).
- Filter & Count: Filter through a 40 μm strainer. Count as in Protocol 1.
Visualized Workflows
Title: Nuclei Extraction Workflow for Challenging Samples
Title: Challenge-Solution-Reagent Mapping for scATAC-seq
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions
| Item | Function & Rationale | Example Product/Catalog |
|---|---|---|
| Nuclei EZ Lysis Buffer | Gentle, hypotonic lysis optimized for nuclei preservation from tissues. Preferable to harsh detergents. | Sigma-Aldrich, NUC101 |
| RNase Inhibitor (Recombinant) | Critical for RNase-rich tissues (pancreas, spleen). Protects nuclear RNA which can be co-assayed. | Protector RNase Inhibitor, 3335402001 |
| Iodixanol (OptiPrep) | Used to create density gradient for debris removal, enhancing nuclei purity without centrifugation damage. | Sigma-Aldrich, D1556 |
| DNase I (Rapidly Dissolvable) | For post-thaw treatment of frozen cells to dissociate DNA-mediated clumps without damaging nuclei. | Worthington, LS002139 |
| Dounce Homogenizer (Glass) | Provides controlled mechanical disruption for fibrous tissues. Tight clearance pestle (B) is key. | Kimble, 885301-0002 |
| 40μm Nylon Mesh Strainers | Removes large debris and unfixed clumps. Pre-wetting with BSA reduces nuclei sticking. | Pluriselect, 43-50040-03 |
| Broad-Spectrum Nuclease Inhibitor | Suppresses endogenous DNase/RNase activity during lysis, especially in spleen/liver. | 10x Genomics, Nuclease Inhibitor (2000088) |
| Propidium Iodide/Acridine Orange (AO/PI) | Dual-fluorescence dye for precise count of intact vs. damaged nuclei on a hemocytometer. | Logos Biosystems, F23001 |
Benchmarking scATAC-seq: Data Validation, Quality Metrics, and Technology Comparison
Within the broader thesis on optimizing the 10x Genomics scATAC-seq workflow and library preparation, three key success metrics emerge as critical for evaluating data quality and biological interpretability: Transcriptional Start Site (TSS) Enrichment, Fragments per Cell, and Peak Calls. These quantitative measures collectively assess library complexity, signal-to-noise ratio, and the effective capture of open chromatin regions, directly influencing downstream analyses in drug discovery and basic research.
Metric Definitions & Benchmark Values
The following table summarizes current benchmark values for high-quality scATAC-seq data generated using the 10x Genomics Chromium platform, as per recent literature and best practice guidelines.
Table 1: scATAC-seq Key Success Metrics and Benchmarks
| Metric | Definition | Minimum Quality Threshold | Target (High Quality) | Interpretation |
|---|---|---|---|---|
| Fragments per Cell | Total number of unique, deduplicated ATAC-seq fragments mapped to the nuclear genome per cell barcode. | > 1,000 fragments | 5,000 - 50,000+ fragments | Measures sequencing depth and library complexity per cell. Low counts indicate poor tagmentation or cell loss. |
| TSS Enrichment | Ratio of fragment density at Transcription Start Sites (± 2kb) to fragment density in flanking regions (± 5kb). | > 3 | 7 - 20+ | Assesses signal-to-noise ratio. Higher values indicate precise, specific cleavage at open chromatin. |
| Peak Calls | Number of non-overlapping, consensus open chromatin regions identified from the aggregate cell population. | N/A (context dependent) | 50,000 - 150,000 peaks | Reflects the breadth of chromatin accessibility captured. Influenced by cell number, diversity, and depth. |
Detailed Protocols for Metric Calculation
Protocol 3.1: Calculating Fragments per Cell and TSS Enrichment with Cell Ranger ATAC
This protocol details the primary data processing using 10x Genomics' official pipeline.
Materials:
- Raw sequencing data (FASTQ files)
cellranger-atac(version 2.1.0 or later)- Reference genome (pre-built for GRCh38/hg38, mm10)
- High-performance computing cluster (recommended)
Procedure:
- Setup: Install
cellranger-atacand download the appropriate reference genome package from the 10x Genomics support site. - Create CSV: Prepare a simple CSV file linking sample names to FASTQ file paths.
- Run Pipeline: Execute the main count pipeline:
- Output Analysis: Upon completion, key metrics are found in:
web_summary.html: Interactive summary.summary.csv: Comma-separated values of all metrics.fragments.tsv.gz: File used for downstream analysis. The "Fragments per Cell" is reported directly. TSS enrichment is calculated internally by aggregating fragment starts around annotated TSSs.
Protocol 3.2: Generating Consensus Peak Calls with MACS2
This protocol describes a common method for creating a consensus peak set from aggregated scATAC-seq fragments.
Materials:
fragments.tsv.gzfile from Cell Ranger ATAC output.- Python environment with
pyatacor tools to convert fragments to BED. - MACS2 (version 2.2.7.1)
- BEDTools suite.
Procedure:
- Fragment Aggregation: Convert the fragment file for all cells into a single, aggregated BED file of fragment ends (representing transposition events).
Peak Calling: Run MACS2 in
--nomodelmode, as ATAC-seq fragments have a fixed size distribution.Filter & Format: Filter peaks for significance and create a final, non-overlapping peak set.
Quantification: Use this
merged_consensus_peaks.bedfile as the reference for creating a peak x cell matrix in downstream tools (e.g., Signac, ArchR).
Visualizations
Diagram 1: scATAC-seq Analysis Workflow & Key Metrics
Diagram 2: TSS Enrichment Score Calculation
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents & Materials for 10x scATAC-seq
| Item | Function in Workflow | Critical for Metric |
|---|---|---|
| Chromium Next GEM Chip K | Partitions single nuclei with gel beads into nanoliter-scale droplets. | Fragments per Cell (efficient cell capture) |
| 10x Barcoded Gel Beads | Contain unique oligonucleotide barcodes for cell-of-origin labeling. | All (defines cellular identity) |
| Tn5 Transposase (Loaded) | Enzyme that simultaneously fragments and tags accessible DNA with adapters. | TSS Enrichment, Peak Calls (specificity of cleavage) |
| Nuclei Buffer | Stabilizes isolated nuclei to prevent lysis and clumping. | Fragments per Cell (preserves integrity) |
| High-Sensitivity DNA Assay Reagents (e.g., Qubit, Bioanalyzer) | Quantify and quality-check pre- and post-amplified libraries. | Fragments per Cell (prevents over/under sequencing) |
| SPRIselect Beads | Perform size selection and clean-up of libraries, removing small fragments. | TSS Enrichment (improves signal-to-noise) |
| Dual Index Kit TT Set A | Provides sample indexes for multiplexing libraries prior to sequencing. | All (enables multiplexing for statistical power) |
| PhiX Control v3 | Spiked into runs for sequencing quality monitoring. | All (ensures base call accuracy for mapping) |
Within the broader research thesis on the 10x Genomics scATAC-seq workflow, validation and integration of chromatin accessibility data with transcriptional outputs are paramount. The 10x Multiome ATAC + Gene Expression assay enables simultaneous profiling of gene expression and chromatin accessibility from the same single nucleus/cell. This Application Note details protocols for multiomic validation, transforming standalone scATAC-seq findings into robust, functionally annotated discoveries.
Table 1: Performance Metrics of 10x Multiome Assay (v1.1)
| Metric | Typical Output | Notes |
|---|---|---|
| Nuclei Recovery Rate | 5,000 - 10,000 nuclei per reaction | Critical for library complexity. |
| Median High-Quality Fragments per Nucleus (ATAC) | 15,000 - 50,000 | Filtering threshold: TSS enrichment >3, fragments in peaks >20%. |
| Median Genes per Nucleus (GEX) | 2,000 - 5,000 | Filtering threshold: 500-5,000 unique genes. |
| Estimated Doublet Rate | 0.8% per 1,000 nuclei | Use DoubletFinder or scrublet for identification. |
| Paired Data Rate | >65% of nuclei passing QC | Nuclei with detectable signal in both modalities. |
| Recommended Sequencing Depth (PE150) | ATAC: 25-50K fragments/nucleus; GEX: 20-50K reads/nucleus | Scales with nucleus target recovery. |
Table 2: Comparison of Single-Modality vs. Multiome Approaches
| Aspect | scATAC-seq Alone | 10x Multiome (ATAC+GEX) |
|---|---|---|
| Data Type | Chromatin accessibility only. | Paired accessibility & expression. |
| Key Analysis | Peak calling, motif enrichment, footprinting. | Direct cis-regulatory link prediction, paired differential analysis. |
| Cell Annotation | Indirect, via gene activity scores. | Direct, via native mRNA expression. |
| Validation Workflow | Requires separate scRNA-seq integration (statistical). | Built-in biological validation from same cell. |
| Cost & Complexity | Lower | Higher, but provides integrated data. |
Detailed Experimental Protocol: Multiome Library Preparation
Principle: The assay uses nuclei isolation, followed by simultaneous transposition of accessible chromatin and capture of mRNA using Gel Bead-In-Emulsions (GEMs). Unique shared barcodes link ATAC and GEX data from the same nucleus.
Materials & Reagents:
- Fresh or frozen tissue/cells.
- Nuclei Buffer (10x Genomics, Cat # 2000153).
- Chromium Next GEM Chip K (Cat # 1000286).
- Chromium Next GEM Multiome ATAC + Gene Expression Kit (Cat # 1000285).
- Dual Index Kit TT Set A (Cat # 1000215).
- SPRIselect Reagent Kit (Beckman Coulter, Cat # B23318).
- Thermal cycler with 53°C lid.
- Magnetic separator for 0.2 mL tubes.
- Agilent 4200 TapeStation or Bioanalyzer.
Procedure:
Part A: Nuclei Isolation & Transposition
- Prepare Nuclei Suspension: Homogenize tissue or dissociate cells. Filter and wash with cold PBS. Lyse cytoplasm using chilled Nuclei Buffer for 5 minutes on ice. Centrifuge, resuspend pellet in chilled Diluted Nuclei Buffer. Count and assess integrity (Trypan Blue). Adjust concentration to 4,000-10,000 nuclei in 10µL.
- Tagmentation: Combine 10µL nuclei, 10µL Tagmentation Buffer, and 5µL Tagmentation Enzyme. Mix gently and incubate at 37°C for 60 minutes. Immediately proceed to GEM generation.
Part B: GEM Generation & Library Construction
- Load Chromium Chip: Combine transposed nuclei, Master Mix, and Gel Beads. Load into a Chromium Next GEM Chip K with partitioning oil. Run on a Chromium Controller to generate ~10,000 GEMs.
- Reverse Transcription & Pre-Amplification: Perform RT in a thermal cycler (53°C for 45 min). Break emulsions, recover post-RT mix, and clean up with SPRIselect beads. Perform a 12-cycle PCR to add P5/P7 handles and sample index (i7) for the GEX library.
- Fragment, Amplify & Index ATAC Library: Treat a portion of the post-RT product with Proteinase K. Perform a 12-cycle PCR to add P5/P7 handles to ATAC fragments. Clean up both GEX and ATAC libraries with SPRIselect beads (0.6x ratio for size selection).
- Final Indexing & QC: Perform a final 8-cycle PCR to add unique dual indices (i5 and i7) to each library. Clean up with SPRIselect beads (0.8x ratio). Quantify using Qubit and profile fragment size (TapeStation). Expected profiles: GEX ~500-6000 bp broad peak; ATAC ~<1000 bp with periodicity.
Diagrams
Title: Multiome Assay Workflow from Nuclei to Data
Title: Multiome Validates cis-Regulatory Links
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Multiome Validation Experiments
| Item | Function | Example (Supplier/Cat #) |
|---|---|---|
| Nuclei Isolation Buffer | Lyse cytoplasmic membrane while preserving nuclear integrity for clean ATAC and RNA signal. | Nuclei Buffer (10x Genomics, #2000153) |
| Tn5 Transposase | Enzyme that simultaneously fragments and tags accessible chromatin with sequencing adapters. | Tagmentation Enzyme (10x Multiome Kit) |
| Chromium Next GEM Chip K | Microfluidic device to partition single nuclei into nanoliter-scale GEMs with barcoded beads. | 10x Genomics (#1000286) |
| Dual Index Oligos | Unique combinatorial indices for multiplexing multiple samples in a single sequencing run. | Dual Index Kit TT Set A (10x, #1000215) |
| SPRIselect Beads | Solid-phase reversible immobilization beads for size selection and cleanup of DNA libraries. | SPRIselect (Beckman Coulter, #B23318) |
| High-Sensitivity DNA/RNA Assay | Accurate quantification and quality control of final libraries prior to sequencing. | Agilent High Sensitivity D1000/RNA ScreenTape (#5067-5584/#5067-5579) |
| Cell Ranger ARC | Primary analysis software for demultiplexing, alignment, and feature counting of paired data. | 10x Genomics Cell Ranger ARC (v2.1.0+) |
| Signac / Seurat | Integrated R toolkit for joint analysis, visualization, and cis-regulatory link prediction. | Signac (v1.10.0) with Seurat (v5.0.0) |
Comparing 10x to Other scATAC Platforms (sci-ATAC-seq, dsciATAC-seq)
Within the broader thesis on optimizing the 10x Genomics scATAC-seq workflow, this analysis provides a critical, comparative framework. Understanding the technical and performance distinctions between the dominant commercial platform (10x Chromium) and key alternative methods (sci-ATAC-seq, dsciATAC-seq) is essential for informed experimental design, data interpretation, and future protocol development. This document serves as an application note, detailing protocols and comparative metrics to guide researchers in selecting the appropriate platform for their specific biological questions and resource constraints.
Platform Principles
- 10x Genomics Chromium scATAC-seq: Utilizes a microfluidic system to partition nuclei into Gel Bead-In-EMulsions (GEMs). Each gel bead is coated with uniquely barcoded oligonucleotides containing a transposase Tn5 adapter sequence. Within each droplet, indexed tagmentation occurs, labeling all chromatin fragments from a single nucleus with a unique cellular barcode.
- sci-ATAC-seq (Single-cell Combinatorial Indexing ATAC-seq): A plate-based, combinatorial indexing approach. Nuclei are first tagmented in bulk with Tn5 transposase loaded with adapters containing a first set of barcodes (i-index). Nuclei are then distributed across a multi-well plate, where a second round of tagmentation or PCR with a second set of barcodes (j-index) occurs. A single cell's fragments are identified by a unique combination of i and j barcodes.
- dsciATAC-seq (Droplet-based Single Cell Combinatorial Indexing ATAC-seq): A hybrid method that combines principles of sci-ATAC-seq with droplet partitioning. Nuclei are pre-indexed (first barcode) in a plate, then pooled and loaded into a droplet-based system (like 10x Chromium) for a second round of barcoding, aiming to increase throughput and reduce costs compared to standard 10x.
Quantitative Comparison Table
Table 1: Comparative Summary of scATAC-seq Platforms
| Feature | 10x Genomics Chromium scATAC-seq | sci-ATAC-seq (v3) | dsciATAC-seq |
|---|---|---|---|
| Partitioning Principle | Microfluidic Droplets (GEMs) | Combinatorial Indexing (Plate-based) | Combinatorial Indexing + Droplets |
| Throughput (Cells/Run) | 500 - 10,000+ (standard) | 10,000 - 100,000+ | 50,000 - 200,000+ (estimated) |
| Cell Multiplexing | No (single barcode per cell) | Yes (dual-index combinatorial) | Yes (dual-index combinatorial) |
| Library Prep Cost per Cell | High (~$0.50 - $1) | Very Low (~$0.05 - $0.10) | Low (~$0.10 - $0.20) |
| Instrumentation | Commercial (Chromium Controller) | Open-source (Lab equipment: multichannel pipettes, thermocyclers) | Hybrid (Lab equipment + Chromium Controller) |
| Hands-on Time | Low (Automated partitioning) | High (Multi-step plate transfers) | Medium-High |
| Data Complexity | Standard (Cell Ranger pipeline) | High (Demultiplexing combinatorial indices) | Very High (Dual demultiplexing) |
| Key Advantage | Robust, standardized, user-friendly workflow | Extremely high throughput at low cost | Ultra-high throughput with reduced background |
| Key Limitation | Cost per cell, fixed throughput per chip | Complex protocol, high doublet rate at high saturation | Highly complex protocol and data processing |
Detailed Experimental Protocols
Core Protocol: Nuclei Isolation from Frozen Tissue (Common to All Platforms)
Reagents: Dounce Homogenizer, Nuclei EZ Lysis Buffer (Sigma NUC-101), 0.4% Trypan Blue, 1x PBS + 0.04% BSA, Protease Inhibitors. Procedure:
- Mince 20-50 mg of frozen tissue on dry ice.
- Transfer to a Dounce homogenizer containing 2 mL of ice-cold Nuclei EZ Lysis Buffer with Protease Inhibitors.
- Homogenize with 10-15 strokes of the loose pestle (A), then 10-15 strokes of the tight pestle (B) on ice.
- Incubate on ice for 5 minutes.
- Filter the lysate through a 40 μm cell strainer into a 15 mL conical tube.
- Centrifuge at 500g for 5 minutes at 4°C to pellet nuclei.
- Gently resuspend the pellet in 2 mL of Lysis Buffer, incubate on ice for 5 min, and centrifuge again (500g, 5 min, 4°C).
- Discard supernatant and gently resuspend nuclei in 1 mL of 1x PBS + 0.04% BSA.
- Filter through a 20 μm strainer. Count using a hemocytometer with Trypan Blue.
- Adjust concentration to the target for the chosen platform (e.g., ~1,000 nuclei/μL for 10x).
Platform-Specific Tagmentation & Library Prep
Protocol 3.2.1: 10x Genomics Chromium scATAC-seq Library Preparation
- Key Reagent: 10x Chromium Next GEM Chip, scATAC-seq Gel Beads, Partitioning Oil.
- Procedure: Follow the manufacturer's "Chromium Next GEM Single Cell ATAC Reagent Kits" user guide (CG000209). Key steps:
- Master Mix Preparation: Combine nuclei, transposase enzyme, and buffer.
- Chip Loading: Load master mix, gel beads, and partitioning oil into the designated wells of the Chromium Next GEM Chip.
- GEM Generation & Barcoding: Run the chip on the Chromium Controller. Within each GEM, transposase tagmentation fragments chromatin, and fragments are linked to the gel bead's unique barcode.
- Post-Processing: Break droplets, purify barcoded DNA, and perform PCR amplification.
- Library Cleanup: Use SPRIselect beads to size-select and clean the final library.
Protocol 3.2.2: sci-ATAC-seq Library Preparation (Simplified Overview)
- Key Reagent: 96-well plate, Tn5 transposase pre-loaded with i7-indexed oligos (i-index), PCR mix with j-index primers.
- Procedure (based on Cusanovich et al., 2018 & 2021):
- Round 1 Tagmentation: Distribute a suspension of nuclei across a 96-well plate (~100-500 nuclei/well). To each well, add Tn5 transposase pre-loaded with a unique i7 index (i-index). Perform tagmentation (37°C, 30-60 min).
- Quenching: Add SDS to each well to quench Tn5 activity.
- Pooling: Pool all tagmented nuclei from all wells into a single tube. Now, each nucleus' fragments carry a well-specific i-index.
- Redistribution: Redistribute the pooled nuclei into a new 96-well PCR plate at a very low density (~1-5 nuclei/well theoretically).
- Round 2 Indexing (PCR): In each new well, perform a limited-cycle PCR using a unique j-index primer pair. This adds a second barcode (j-index) to fragments.
- Final Library: Pool all PCR products from the second plate. A true single-cell library has a unique combination of one i-index and one j-index.
Protocol 3.2.3: dsciATAC-seq Library Preparation (Hybrid Workflow)
- Key Reagent: 96-well plate for pre-indexing, 10x Chromium system for second indexing.
- Procedure (based on Lareau et al., Nature Biotech 2021):
- Pre-Tagmentation (Plate-based): Perform a first round of limited, indexed tagmentation in a 96-well or 384-well plate, similar to sci-ATAC-seq Step 1. This attaches the first barcode (i-index).
- Pool and Quench: Pool all pre-tagmented nuclei and quench the reaction.
- Droplet Partitioning (10x): Load the pooled, pre-indexed nuclei onto the 10x Chromium system using a standard 10x scATAC-seq v2 kit, but omit the transposase enzyme from the master mix. The gel beads provide the second barcode (j-index) during in-droplet PCR.
- Library Generation: Complete the 10x protocol from post-GEM generation through library amplification. The final library contains dually-indexed fragments.
Signaling Pathways & Workflow Visualizations
Diagram 1: Core Workflow Comparison of 10x vs sci-ATAC-seq
Diagram 2: dsciATAC-seq Hybrid Method Steps
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for scATAC-seq Experiments
| Reagent / Material | Function | Platform Specificity |
|---|---|---|
| Chromium Next GEM Chip H | Microfluidic chip to generate Gel Bead-In-EMulsions (GEMs) for single-cell partitioning. | 10x Genomics (essential) |
| 10x scATAC-seq v2 Gel Beads | Beads containing unique barcodes and Tn5 adapter sequences for in-droplet barcoding. | 10x Genomics (essential) |
| Custom Tn5 Transposase | Enzyme loaded with specific indexed adapter oligos for targeted tagmentation. | sci-ATAC-seq, dsciATAC-seq (essential) |
| Nuclei EZ Lysis Buffer | Gentle, non-ionic detergent buffer for isolating clean nuclei from tissues/cells. | Universal (common) |
| SPRIselect Beads | Solid-phase reversible immobilization beads for DNA size selection and cleanup. | Universal (common) |
| Reduced-EDTA TE Buffer | Elution/storage buffer that preserves chromatin integrity and prevents over-tagmentation. | Universal (recommended) |
| Droplet Generation Oil | Oil for creating stable microfluidic emulsions on the Chromium system. | 10x Genomics, dsciATAC-seq (essential) |
| 384-Well LoBind Plates | Low-adhesion plates for minimizing nucleus loss during combinatorial indexing steps. | sci-ATAC-seq, dsciATAC-seq (recommended) |
| PCR Reagents (dNTPs, Polymerase) | For amplifying barcoded libraries post-tagmentation. | Universal (essential) |
| Dual Index Kit Set A (SI-PCR) | Provides unique i5/i7 index primers for final library multiplexing for sequencing. | Universal (essential, format varies) |
This application note details a framework for validating candidate regulatory elements and gene targets identified through single-cell ATAC-seq (scATAC-seq) analysis, specifically within a research thesis focused on optimizing the 10x Genomics Chromium scATAC-seq workflow and library preparation. The 10x Genomics platform enables the generation of chromatin accessibility profiles from thousands of individual cells, yielding maps of putative cis-regulatory elements (cREs) and inferred gene-regulatory networks. However, these computational inferences require empirical validation through functional assays to confirm biological relevance, especially for downstream applications in target discovery and drug development.
Key Quantitative Data from scATAC-seq Analysis
Table 1: Summary Statistics from a Representative scATAC-seq Experiment (10x Genomics Platform)
| Metric | Value | Description |
|---|---|---|
| Cells Recovered | 12,458 | Number of cell barcodes after filtering. |
| Median Fragments per Cell | 78,432 | Median sequencing fragments per cell. |
| Fraction of Fragments in Peaks | 42% | Proportion of fragments overlapping consensus peak set. |
| TSS Enrichment Score | 18.7 | Measure of signal-to-noise at transcription start sites. |
| Non-Redundant Fragments (NRF) | 68% | Fraction of unique fragments (PCR duplicate removal). |
| Identified Cell Clusters | 12 | Distinct cell states identified via latent semantic indexing. |
| Candidate cREs | 45,201 | Accessible regions called across all clusters (FDR < 0.01). |
| Linked Candidate Target Genes | 3,842 | Genes associated with cREs via correlation or linkage. |
Table 2: Prioritized Candidate Genes for Functional Validation
| Gene Symbol | scATAC-seq Cluster | -log10(P-value) | Putative Function | Assay Planned |
|---|---|---|---|---|
| MYC | Proliferating | 12.5 | Transcription factor, proliferation | CRISPRi, RT-qPCR |
| IL2RA | Treg | 9.8 | Immune regulation, receptor | Luciferase, Flow Cytometry |
| SOX9 | Progenitor | 11.2 | Developmental regulator | CRISPRa, scRNA-seq |
| PDL1 (CD274) | Exhausted | 8.3 | Immune checkpoint | Luciferase, Cytokine Assay |
Detailed Experimental Protocols
Protocol 3.1: Candidate Enhancer Validation via Luciferase Reporter Assay
Objective: To test the transcriptional activity of candidate regulatory elements (cREs) identified from scATAC-seq peaks. Materials: pGL4.23[luc2/minP] vector, Restriction enzymes, Kapa Hifi HotStart ReadyMix, K562 or HEK293T cells, Lipofectamine 3000, Dual-Luciferase Reporter Assay System. Procedure:
- Amplify and Clone cREs: Design primers with overhangs for In-Fusion cloning to amplify ~500-1000bp genomic regions centered on the scATAC-seq peak summit from genomic DNA. Gel-purify PCR products.
- Vector Preparation: Linearize the pGL4.23 vector using appropriate restriction enzymes (e.g., KpnI and NcoI). Purify the digested vector.
- Cloning: Perform In-Fusion reaction to insert the purified PCR product into the linearized vector. Transform into Stbl3 competent cells. Sequence-validate multiple clones.
- Cell Transfection: Seed 2 x 10^4 HEK293T cells per well in a 96-well plate. The next day, co-transfect 100ng of experimental firefly luciferase reporter construct and 10ng of pRL-SV40 Renilla luciferase control vector using Lipofectamine 3000 per manufacturer's protocol.
- Luciferase Measurement: 48 hours post-transfection, lyse cells and measure firefly and Renilla luciferase activity sequentially using the Dual-Luciferase Reporter Assay System on a plate reader.
- Data Analysis: Normalize firefly luciferase activity to Renilla activity for transfection efficiency. Compare activity of the cRE construct to the empty vector (minimal promoter only) control. Perform experiments in biological triplicates.
Protocol 3.2: Functional Validation of Target Genes via CRISPRi/a
Objective: To perturb the expression of a candidate target gene linked to a validated cRE and assess phenotypic consequences. Materials: dCas9-KRAB (CRISPRi) or dCas9-VPR (CRISPRa) expressing cell line, lentiviral sgRNA vectors, Puromycin, TRIzol, RT-qPCR kit, relevant phenotypic assay reagents. Procedure:
- sgRNA Design: Design 3-5 sgRNAs targeting the transcriptional start site (TSS) of the candidate gene for CRISPRi/a, or within the validated cRE for CRISPRi-mediated silencing of the element itself. Use a non-targeting sgRNA as control. Clone into a lentiviral sgRNA expression vector (e.g., lentiGuide-Puro).
- Virus Production and Transduction: Produce lentivirus in Lenti-X 293T cells using psPAX2 and pMD2.G packaging plasmids. Transduce target cells (expressing dCas9 effector) with viral supernatant plus polybrene (8µg/mL). Select with puromycin (1-2µg/mL) for 72 hours starting 48h post-transduction.
- Validation of Gene Perturbation: Harvest RNA from pelleted cells using TRIzol. Synthesize cDNA and perform RT-qPCR with primers for the target gene and housekeeping controls (e.g., GAPDH, ACTB). Calculate fold-change relative to non-targeting sgRNA control.
- Phenotypic Assessment: Depending on the predicted gene function, perform relevant assays (e.g., flow cytometry for surface markers, Incucyte monitoring for proliferation, ELISA for cytokine secretion) on the perturbed cell population. Compare to control.
Visualization of Workflows and Pathways
Diagram 1: scATAC-seq to Functional Validation Workflow
Title: scATAC-seq Validation Workflow
Diagram 2: CRISPRi Mechanism for cRE Validation
Title: CRISPRi Silences Enhancer to Validate Function
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Validation Studies
| Reagent/Solution | Vendor Examples | Function in Validation |
|---|---|---|
| 10x Genomics ChromiumNext GEM ATAC Solution | 10x Genomics | Library preparation for generating initial scATAC-seq data. |
| Dual-Luciferase ReporterAssay System | Promega | Quantifies transcriptional activity of cloned candidate cREs. |
| In-Fusion HD Cloning Kit | Takara Bio | Efficient, seamless cloning of PCR-amplified cREs into reporter vectors. |
| lentiGuide-Puro & lenti-dCas9Vectors | Addgene | Lentiviral delivery of sgRNAs and dCas9 effectors for CRISPRi/a. |
| Lipofectamine 3000 | Thermo Fisher | High-efficiency transfection reagent for plasmid delivery. |
| TRIzol Reagent | Thermo Fisher | Simultaneous extraction of RNA, DNA, and protein from cells post-perturbation. |
| Cell Staining Antibodies(e.g., anti-IL2RA, anti-PDL1) | BioLegend | Phenotypic validation of target gene modulation via flow cytometry. |
| Kapa Hifi HotStartReadyMix | Roche | High-fidelity PCR for amplification of genomic regions with minimal error. |
Cost-Benefit and Throughput Analysis for Project Scaling
1. Application Notes
Scaling a 10x Genomics scATAC-seq project from pilot to production necessitates a rigorous cost-benefit and throughput analysis. This evaluation is critical for optimizing resource allocation, ensuring data consistency, and maximizing the scientific return on investment, particularly in drug development where identifying rare cell populations and regulatory landscapes is paramount.
1.1. Key Scaling Decision Factors The decision to scale involves balancing fixed and variable costs against the value of increased cellular throughput and statistical power. A primary consideration is the transition from the Chromium Controller to the Chromium Connect, which introduces automation for increased reproducibility and hands-free operation. The choice between the Chromium X Series and the newer Chromium GEM-X Series chips impacts both cell recovery and per-sample cost.
1.2. Quantitative Scaling Analysis Based on current list prices and published performance specifications (10x Genomics, 2024), the following comparative analysis was constructed.
Table 1: Cost-Benefit Analysis of scATAC-seq Workflow Scaling
| Component | Pilot Scale (Manual) | Production Scale (Automated) | Notes |
|---|---|---|---|
| Instrument | Chromium Controller | Chromium Connect | Connect enables walkaway automation, reducing hands-on time by ~70%. |
| Chip Type | Chromium X | Chromium GEM-X | GEM-X offers higher cell recovery (up to 20% improvement). |
| Max Samples/Run | 8 (with 2 chips) | 16 (with 4 chips on Connect) | Throughput is limited by thermal cycler capacity for library prep. |
| Estimated Hands-On Time | ~6 hours (library prep) | ~2 hours (library prep) | Automation significantly reduces technician time and variability. |
| Reagent Cost per 10k Cells | ~$2,500 | ~$2,200 | Bulk purchasing and reduced waste lower per-cell cost at scale. |
| Primary Benefit | Flexibility, lower initial investment | High throughput, reproducibility, lower per-sample cost |
Table 2: Throughput and Yield Comparison by Chip Type
| Chip Type | Target Cell Recovery | Nuclei Input Range | Recommended Libraries per Chip | Approx. Sequencing Depth per Cell |
|---|---|---|---|---|
| Chromium X | 5,000 - 10,000 | 10,000 - 20,000 | 1 | 25,000 - 50,000 fragments |
| Chromium GEM-X | 6,000 - 12,000 | 12,000 - 24,000 | 1 | 25,000 - 50,000 fragments |
| Chromium GEM-X Dual | 10,000 - 20,000 (total) | 20,000 - 40,000 | 2 | 25,000 - 50,000 fragments |
2. Experimental Protocols
2.1. Protocol: Scalable Nuclei Isolation from Frozen Tissue for scATAC-seq This standardized protocol is optimized for scaling across hundreds of samples with minimal batch effect.
Materials:
- Frozen tissue sample
- Homogenization Buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1% Nonidet P40 Substitute, 1% BSA, 1mM DTT, 1x Protease Inhibitor)
- Wash Buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, 1% BSA, 1mM DTT)
- Nuclei Buffer (10x Genomics Buffer DK, or equivalent: 10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, 1% BSA, 0.1% Tween-20)
- DAPI stain (1 µg/mL) and flow sorter or Countess Cell Counter
- 40µm flowmi cell strainers
- Refrigerated centrifuge
Procedure:
- Homogenize: Place ~25 mg frozen tissue in 1 mL ice-cold Homogenization Buffer in a Dounce homogenizer. Perform 15-20 strokes with the loose pestle (A), then 15-20 strokes with the tight pestle (B). Keep on ice.
- Filter & Lyse: Filter the homogenate through a pre-wet 40µm strainer into a 2mL tube. Incubate on ice for 5 minutes to complete lysis.
- Wash Nuclei: Pellet nuclei at 500 rcf for 5 minutes at 4°C. Carefully aspirate supernatant.
- Resuspend & Filter: Resuspend the pellet in 1 mL Wash Buffer by gentle pipetting. Pellet again at 500 rcf for 5 minutes at 4°C.
- Count & Adjust: Aspirate supernatant. Resuspend nuclei in 100-200 µL Nuclei Buffer. Count using DAPI stain and a hemocytometer or automated counter. Adjust concentration to 1,000-2,000 nuclei/µL for 10x loading. Keep on ice until loading onto the chip.
2.2. Protocol: Automated Library Preparation on Chromium Connect This protocol follows the 10x Genomics Chromium Next GEM Chip and Library Kit user guide (CG000492 Rev C), highlighting scaling parameters.
Materials:
- Chromium Connect instrument
- Chromium Next GEM Chip GEM-X (Dual)
- Chromium Next GEM scATAC-seq Reagent Kit v2
- High Sensitivity D1000 ScreenTape (Agilent) or FEMTO Pulse
- SPRIselect beads (Beckman Coulter)
- Thermal cycler with 96-well block
Procedure:
- Instrument Setup: Prime the Chromium Connect as per manufacturer's instructions. Load the required reagents (Master Mix, Partitioning Oil, Gel Beads) into the designated cartridge slots.
- Sample Plate Preparation: In a hard-shell 96-well plate, combine up to 16 samples of nuclei suspension (adjusted to 1,000-2,000 nuclei/µL) with the provided Master Mix. Seal the plate.
- Chip Loading & Run: Load the sample plate and up to 4 Chromium Next GEM Chip GEM-X (Dual) chips into the instrument. Start the automated run. The system will perform nuclei/GEM partitioning, barcoding, and post-GEM cleanup, outputting purified cDNA in a 96-well plate.
- Scalable Library Construction: Transfer the cDNA plate to a thermal cycler. Perform the indexing PCR as per the kit protocol. Pool reactions as needed based on experimental design.
- Library Cleanup & QC: Perform a double-sided SPRIselect bead cleanup (0.55x and 0.9x ratios) to size-select the final library. Quantify using a high-sensitivity, long-fragment assay (e.g., Agilent High Sensitivity D1000). Libraries should show a broad smear from ~300bp to >1,000bp.
3. Mandatory Visualizations
Title: Decision Pathway for Scaling scATAC-seq Projects
Title: High-Throughput scATAC-seq Experimental Workflow
4. The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Scalable scATAC-seq Research
| Reagent / Material | Function in Workflow | Key Consideration for Scaling |
|---|---|---|
| Chromium Next GEM Chip GEM-X (Dual) | Microfluidic device for partitioning single nuclei into Gel Bead-in-Emulsions (GEMs). | Dual sample capability doubles throughput per chip, reducing per-sample cost and instrument run time. |
| Chromium Next GEM scATAC-seq v2 Reagent Kit | Contains all enzymes, buffers, and primers for tagmentation, barcoding, and library construction. | Bulk purchasing for production scale is essential. Kit stability ensures consistency across large batches. |
| Buffer DK (Nuclei Buffer) | Optimized buffer for nuclei suspension and loading onto the chip. Maintains nuclear integrity and prevents clumping. | Critical for achieving consistent nuclei recovery across many samples. Can be prepared in large, QC'd batches. |
| SPRIselect Beads (Beckman Coulter) | Magnetic beads for size-selective purification of post-GEM cDNA and final libraries. | Enables rapid, high-throughput cleanup in 96-well plates. Ratio optimization is crucial for fragment selection. |
| High Sensitivity D1000 ScreenTape (Agilent) | Used for quantitative and qualitative QC of final libraries prior to sequencing. | Automated tape station analysis (e.g., 4200 TapeStation) allows rapid, standardized QC of hundreds of libraries. |
| Unique Dual Index Kit, 96 UDIs | Provides 96 unique combinatorial indexes for multiplexing samples on a sequencing run. | Essential for pooling many libraries without index collision, maximizing sequencing lane efficiency. |
Conclusion
The 10x Genomics scATAC-seq workflow provides a robust, scalable, and user-friendly pipeline for constructing high-resolution maps of the regulatory genome across thousands of individual cells. Mastery of the foundational principles, meticulous execution of the library preparation protocol, proactive troubleshooting, and rigorous validation against established metrics are all critical for generating biologically impactful data. As the field advances, the integration of scATAC-seq with transcriptomic, proteomic, and spatial technologies promises to unlock comprehensive models of gene regulation. This will profoundly accelerate discovery in fundamental biology, the identification of novel therapeutic targets, and the development of personalized diagnostic strategies in cancer, immune disorders, and neurodevelopmental diseases.