Optimized Protocol for Nuclei Isolation from Frozen Postmortem Brain Tissue: A Guide for Single-Cell Omics
This article provides a comprehensive guide for researchers and drug development professionals on isolating high-quality nuclei from frozen postmortem brain tissue, a critical step for single-nuclei RNA sequencing (snRNA-seq) and...
Optimized Protocol for Nuclei Isolation from Frozen Postmortem Brain Tissue: A Guide for Single-Cell Omics
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on isolating high-quality nuclei from frozen postmortem brain tissue, a critical step for single-nuclei RNA sequencing (snRNA-seq) and other genomic applications. We cover the foundational principles of overcoming cellular heterogeneity, present detailed methodological protocols including fluorescence-activated nuclei sorting (FANS), address common troubleshooting and optimization challenges specific to low-input and precious samples, and discuss validation techniques to ensure data quality and comparability with existing single-cell atlases. This resource is designed to maximize the research potential of valuable biobanked brain specimens.
Why Isolate Nuclei? Overcoming the Challenges of Postmortem Brain Tissue
The Problem of Cellular Heterogeneity in the Brain
The human brain represents a pinnacle of biological complexity, comprising a vast and diverse array of cell types that orchestrate cognition, behavior, and neural homeostasis. This cellular diversity includes not only broad categories such as neurons, astrocytes, oligodendrocytes, and microglia but also numerous subtypes within these categories with distinct molecular signatures and functions [1]. The cerebral cortex alone contains approximately 16.3 billion neurons [1], far surpassing the neuronal counts of closely related species and highlighting the extraordinary cellular complexity of the human brain.
This remarkable heterogeneity, while fundamental to brain function, presents significant challenges for research. Traditional bulk analysis methods, which combine signal across all cell types, obscure critical cell-type-specific changes in gene expression, epigenetic modifications, and molecular responses to disease [2]. As the field moves toward understanding neurological and psychiatric disorders at a mechanistic level, overcoming this analytical limitation has become imperative. Single-nucleus genomics has emerged as a powerful solution, particularly when applied to frozen postmortem brain tissue, allowing researchers to deconvolve this complexity by analyzing individual nuclei from previously inaccessible sample types [2] [3] [1].
Nuclei Isolation from Frozen Postmortem Brain Tissue: Methodological Considerations
Advantages of Single-Nucleus Approaches
The use of nuclei rather than whole cells for single-cell analyses offers several distinct advantages for brain research, especially when working with frozen postmortem tissue. Unlike whole-cell RNA sequencing, which requires fresh tissue and is susceptible to dissociation-induced stress responses, single-nucleus RNA sequencing (snRNA-seq) enables the profiling of archived clinical materials from brain banks [1]. This approach effectively decouples tissue acquisition from immediate processing, a significant logistical advantage for clinical studies [4].
Critically, nuclei isolation minimizes the cell-type dissociation biases that often plague whole-cell preparations, particularly for fragile cell types like neurons [3]. While nuclei datasets may lack certain cytoplasmic transcripts, they consistently replicate findings from single-cell studies and in some cases provide a more accurate representation of in vivo cellular composition [4] [5]. This technical advantage makes snRNA-seq particularly valuable for creating comprehensive brain cell atlases and studying complex neurological disorders where postmortem tissue is the primary available material.
Technical Challenges and Optimization Needs
Isolating nuclei from frozen postmortem primate brain tissue presents unique technical hurdles that require specialized protocols. These tissues often exhibit reduced RNA integrity due to postmortem intervals and elevated levels of myelin debris, particularly in primates with their higher proportion of white matter compared to rodent models [2]. Additionally, the physical properties of frozen brain tissue necessitate optimization of homogenization techniques to balance nuclei yield against preservation of nuclear integrity.
Existing standard protocols often require substantial starting material (several hundred milligrams) that may exceed the size of smaller brain regions of interest [2]. They may also depend on specialized equipment like ultracentrifuges that are not universally available [2]. Commercial kits developed for general use frequently prove suboptimal for the particular challenges of frozen postmortem brain tissue, resulting in reduced nuclei integrity, limited resolution during flow cytometry, and elevated debris levels that compromise sorting efficiency [2]. These limitations underscore the need for optimized, brain-specific nuclei isolation methods that address the unique characteristics of neural tissue.
Comparative Analysis of Nuclei Isolation Methods
To guide method selection, we systematically evaluated three distinct nuclei isolation strategies using mouse brain cortex tissue, assessing their performance across multiple quantitative metrics. Each method was tested with approximately 30mg of input tissue to enable direct comparison [3].
Table 1: Performance Comparison of Nuclei Isolation Methods
| Method | Nuclei Yield (per mg tissue) | Nuclei Integrity | Debris Level | Cell Type Bias | Equipment Needs |
|---|---|---|---|---|---|
| Sucrose Gradient Centrifugation | ~60,000 | 85% | Minimal | Higher astrocyte proportion (13.9%) | Ultracentrifuge required |
| Spin Column-Based | 25% less than other methods | 35% | Substantial aggregation and debris | Reduced diversity | Specialized columns |
| Machine-Assisted Platform | ~60,000 | ~100% | Negligible | Higher microglia (5.6%) and oligodendrocytes (15.9%) | Specialized automated equipment |
Impact on Downstream Transcriptomic Analyses
The choice of isolation method profoundly influences downstream snRNA-seq data quality and biological interpretation. Beyond the quantitative metrics shown in Table 1, each protocol demonstrates distinct strengths in preserving specific aspects of brain cellular diversity:
- Centrifugation-based methods effectively capture defined individual nuclei with minimal background debris, supporting high-quality transcriptomic data [3].
- Column-based approaches tend to produce densely packed nuclei with notable aggregation, compromising data quality despite successful capture of diverse cell types [3].
- Machine-assisted platforms yield well-separated, intact nuclei with negligible debris, optimizing nuclear integrity but requiring significant equipment investment [3].
These methodological differences directly impact the detection of cell-type-specific markers and introduce protocol-dependent variability in population homogeneity as measured by metrics like the ratio of global unshifted entropy (ROGUE) [3]. Such variations highlight the critical importance of method selection based on specific research goals and the cell populations of primary interest.
Optimized Protocol for Frozen Postmortem Primate Brain Tissue
Specialized Protocol for Challenging Samples
Building on the comparative analysis, we present an optimized protocol specifically tailored to the challenges of frozen postmortem nonhuman primate (NHP) brain tissue, with particular application to chimpanzee cerebral cortex [2]. This protocol addresses the key limitations of standard approaches by modifying lysis conditions, enhancing filtration, and incorporating additional wash steps to improve nuclei yield and integrity while reducing debris from small starting amounts of tissue (~25mg) [2].
Table 2: Key Reagents and Equipment for Optimized Nuclei Isolation
| Item | Function | Specific Example |
|---|---|---|
| Nuclei Isolation Kit | Core isolation reagents | 10X Genomics Chromium Nuclei Isolation Kit with RNase Inhibitor |
| Homogenization Device | Tissue disruption | Dounce homogenizer with loose/clear pestles |
| Filtration System | Debris removal | Flowmi cell strainer (70μm) or MACS strainers (30μm) |
| Centrifugation Media | Nuclei purification | Iodixanol gradient solutions |
| Viability Stain | Nuclei quality assessment | Invitrogen ReadyCount Red/Green Viability Stain |
| Antibodies | Cell type enrichment | Anti-NeuN for neuronal nuclei |
| Fluorescent-Activated Sorter | Nuclei sorting | Sony SH800Z Cell Sorter or BD FACSAria Fusion |
Step-by-Step Protocol Workflow
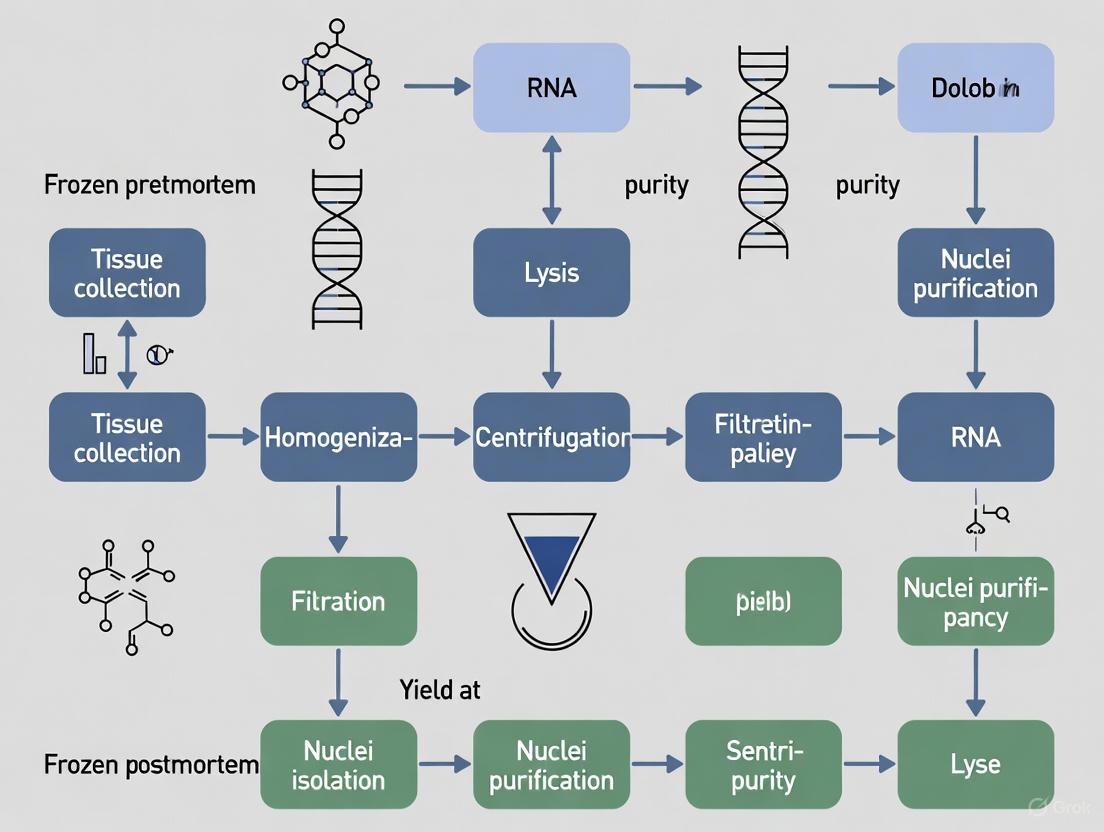
The following workflow diagram illustrates the optimized nuclei isolation and processing procedure:
Detailed Procedural Steps:
Tissue Preparation: Perform microdissection of frozen cerebral cortex using a 2mm biopsy punch on dry ice, weighing ~25-50mg of tissue per reaction [2]. Transfer to pre-chilled microcentrifuge tubes maintained on dry ice throughout the process.
Nuclei Isolation: Use the Chromium Nuclei Isolation Kit with optimized lysis time and increased wash steps. Add a filtration step using a Flowmi cell strainer (70μm) or similar to remove large debris particles [2] [6].
Gradient Purification: Employ a discontinuous iodixanol gradient centrifugation at 1480×g for 20 minutes in a swinging bucket centrifuge with the brake off. Collect the nuclei band located at the interface between 30% and 40% iodixanol [6] [5].
Quantification and Viability Assessment: Combine 5μL of nuclei suspension with 5μL Invitrogen ReadyCount Red/Green Viability Stain. Count using an automated cell counter with GFP and RFP cubes. Successful isolation typically yields a majority of double-stained nuclei [2].
Optional Immunostaining for Neuronal Nuclei: For studies requiring neuronal enrichment, incubate nuclei with primary antibody for NeuN (Neuronal Nuclei) for 30 minutes on ice. Remove unbound antibody by centrifugation and add secondary antibody for 15 minutes in the dark on ice [2].
Fluorescent-Activated Nuclei Sorting (FANS): Analyze samples on a cell sorter (e.g., Sony SH800Z). Use unstained controls to establish voltage settings and negative gates. Add DAPI to identify intact nuclei and collect NeuN-positive and NeuN-negative populations for downstream applications [2].
Downstream Applications and Validation
The isolated nuclei are suitable for various genomic applications, each with specific considerations:
- Single-nucleus RNA-seq: Input nuclei directly into 10X Genomics library preparation protocols without additional sorting. This approach captures the full cellular diversity of the tissue [2] [7].
- Bulk Epigenomic Analyses: Use FANS-enriched neuronal nuclei for methylome sequencing. For low DNA yields from sorted nuclei, employ kits optimized for small inputs (e.g., QIAamp DNA Micro Kit) [2].
- ATAC-seq: Process isolated nuclei for assay for transposase-accessible chromatin sequencing to profile chromatin accessibility in specific brain cell types [2].
Validation of the isolation and sorting efficiency can be performed by sequencing 10X 3'-RNA-seq libraries from both NeuN-positive and NeuN-negative populations, confirming enrichment of neuronal markers in the positive fraction and their absence in the negative fraction [2]. For methylome sequencing from sorted neuronal nuclei, enzymatic conversion methods (e.g., NEBNext Enzymatic Methyl-seq Kit) are recommended over bisulfite conversion due to reduced DNA damage, particularly valuable for low-input samples [2].
Troubleshooting and Quality Control Considerations
Successful implementation of the protocol requires careful attention to potential pitfalls and quality metrics throughout the process. The following diagram outlines key decision points in the quality control pipeline:
Essential Quality Control Metrics:
- Nuclei Integrity: Assess via brightfield microscopy and viability staining. Target >85% intact nuclei for centrifugation-based methods and nearly 100% for machine-assisted platforms [3].
- Debris Contamination: Monitor during filtration and washing steps. High debris levels may require additional filtration through 30μm strainers or optimization of homogenization intensity [2] [7].
- Sorting Efficiency: When using FANS, establish appropriate gates using unstained and secondary antibody-only controls. DAPI staining helps identify intact nuclei [2].
- Downstream QC: For snRNA-seq, ensure nuclei suspensions meet sample input requirements (e.g., 100,000-400,000 nuclei for 10X Genomics) [7]. Post-sequencing, apply computational filters for unique molecular identifiers (UMIs), genes detected per nucleus, and mitochondrial percentage [5].
The problem of cellular heterogeneity in the brain, while presenting significant analytical challenges, can be effectively addressed through optimized nuclei isolation methods that preserve cellular diversity and enable cell-type-specific molecular profiling. The protocol presented here provides a validated approach for extracting high-quality nuclei from frozen postmortem brain tissue, particularly valuable for rare nonhuman primate specimens and human clinical samples.
As single-cell technologies continue to advance, integrating transcriptomic data with other modalities such as epigenomics, proteomics, and spatial information will further enhance our understanding of brain function and dysfunction. The methodology outlined establishes a robust foundation for these integrative approaches, supporting the next generation of discoveries in neuroscience and the development of targeted therapies for neurological and psychiatric disorders.
Advantages of Nuclei over Whole Cells for Frozen Archival Tissue
Single-nucleus RNA sequencing (snRNA-seq) has emerged as a transformative technology for studying complex tissues, particularly when utilizing frozen archival specimens such as postmortem brain tissue. This approach overcomes fundamental limitations associated with traditional single-cell RNA sequencing (scRNA-seq) that relies on intact whole cells. For researchers investigating neurological disorders, neurodegenerative diseases, and brain development, nuclei isolation enables the utilization of valuable biobank resources that were previously challenging to analyze. This application note details the specific advantages of nuclei-based approaches, provides quantitative comparisons of isolation methodologies, and outlines standardized protocols for generating high-quality data from frozen archival brain tissues.
Complex tissues like the brain present significant challenges for single-cell transcriptomic analysis due to their intricate cellular heterogeneity and the fragility of certain cell populations. Traditional single-cell RNA sequencing requires fresh tissue and enzymatic dissociation to create single-cell suspensions, processes that are particularly detrimental to neuronal cells and can activate stress responses in glial populations [3]. These technical artifacts compromise data quality and can skew biological interpretations. Furthermore, the requirement for fresh tissue excludes the vast majority of archived clinical and postmortem specimens from analysis.
Nuclei isolation circumvents these limitations by utilizing the stable nuclear compartment of cells rather than intact whole cells. This paradigm shift enables researchers to:
- Utilize frozen archival tissues including postmortem human brain samples and clinical biopsies stored in biobanks [3] [8].
- Minimize dissociation-induced artifacts that disproportionately affect fragile neuronal populations and activate glial cells [3] [9].
- Preserve cellular diversity by providing more balanced representation of hard-to-isolate cell types like excitatory neurons [3].
- Enable multi-omic profiling of archived specimens for genomic, epigenomic, and transcriptomic analysis from the same sample [8].
Comparative Analysis of Nuclei Isolation Methodologies
Quantitative Performance Metrics Across Isolation Methods
A systematic comparison of three mechanistically distinct nuclei isolation protocols for brain tissue revealed significant differences in performance characteristics critical for experimental success [3].
Table 1: Performance Comparison of Nuclei Isolation Methods for Mouse Brain Cortex
| Method | Nuclei Yield (per mg tissue) | Intact Nuclei (%) | Debris Level | Key Cell Type Proportions |
|---|---|---|---|---|
| Sucrose Gradient Centrifugation | ~60,000 | 85% | Minimal | Highest astrocytes (13.9%) |
| Spin Column-Based | 25% lower yield | 35% | Substantial | Not specified |
| Machine-Assisted Platform | ~60,000 | ~100% | Negligible | Highest microglia (5.6%) and oligodendrocytes (15.9%) |
Impact on Downstream Transcriptomic Data Quality
The choice of isolation method significantly influences snRNA-seq data quality and biological interpretation [3]:
- Centrifugation-based methods produce defined individual nuclei with minimal background debris, facilitating accurate cell type identification.
- Column-based methods often result in nuclei aggregation and substantial debris contamination despite multiple optimization attempts.
- Machine-assisted platforms yield well-separated, intact nuclei with negligible debris, maximizing data quality and reducing ambient RNA contamination.
Different isolation workflows differentially influence contamination levels from ambient, mitochondrial, and ribosomal RNAs, all of which can create technical artifacts that mask true biological effects [3].
Detailed Experimental Protocol for Nuclei Isolation from Frozen Brain Tissue
Two-Day Protocol for Cell Type-Specific Nuclei Isolation
This optimized protocol enables isolation of nuclei from neurons, microglia, oligodendrocytes, and astrocytes from frozen human and rodent brain tissue for downstream omic applications [8].
Day 1: Tissue Preparation and Homogenization
- Tissue Aliquoting: Divide frozen brain tissue into ~150 mg aliquots on dry ice to prevent thawing and maintain nuclei integrity, especially for sensitive cell types like microglia [8].
- Homogenization and Fixation: Homogenize tissue in phosphate buffered saline (PBS) containing 1% (wt/vol) formaldehyde to preserve nuclear epitopes and chromatin structure.
- Fixation Quench: Add glycine to halt formaldehyde fixation and prevent over-fixation.
Day 2: Nuclei Extraction and Staining
- Homogenate Washes: Centrifuge homogenates and carefully remove supernatant to eliminate fixation reagents.
- Mechanical Disruption: Dounce homogenate with loose and tight pestles to liberate nuclei from cellular material.
- Sucrose Cushion Centrifugation: Layer homogenate over sucrose solution to separate nuclei from myelin and cellular debris. The sucrose molarity represents a compromise between debris removal and nuclei yield that may require optimization for specific tissue types [8].
- Antibody Staining: Immunostain nuclei overnight (up to two nights) at 4°C with cell type-specific nuclear markers:
- NeuN for neuronal nuclei
- PU.1 for microglia nuclei
- OLIG2 for oligodendrocyte nuclei
- LHX2 for astrocyte nuclei (gated as NeuN-negative, LHX2-positive) [8]
- Fluorescence-Activated Nuclei Sorting (FANS): Sort immunolabeled single nuclei using DAPI staining and fluorescence-activated sorting with appropriate laser configurations and filter sets for the selected antibody panel.
Critical Protocol Optimization Considerations
- Fixation Methods: While 1% formaldehyde is adequate for most histone modification ChIP-seq, transcription factor binding studies may require double fixation with disuccinimidyl glutarate (DSG) followed by formaldehyde [8].
- Sucrose Cushion Stringency: Test a range of sucrose molarities to optimize the balance between debris removal and nuclei yield for specific tissue sources [8].
- Antibody Panel Design: When expanding antibody panels, include proper isotype controls, adjust for spectral overlap, and verify flow cytometer laser compatibility [8].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Essential Reagents for Nuclei Isolation from Frozen Brain Tissue
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| Fixation Agents | Formaldehyde (1% wt/vol), DSG (for transcription factors) | Preserves nuclear architecture and protein-DNA interactions [8] |
| Separation Media | Sucrose cushion solutions | Separates nuclei from myelin and cellular debris [3] [8] |
| Antibody Panels | NeuN, PU.1, OLIG2, LHX2 | Cell type-specific nuclei identification and sorting [8] |
| Sorting Reagents | DAPI nuclear stain, Fluorescent secondary antibodies | Nuclei visualization and fluorescence-activated sorting [8] |
| Commercial Platforms | 10x Genomics Chromium, Machine-assisted isolation systems | High-throughput nuclei processing and library preparation [3] [10] |
Applications and Integration with Multi-Omic Approaches
Nuclei isolation from frozen archival brain tissue enables diverse downstream genomic and epigenomic applications that are transforming our understanding of neurological disorders:
- Epigenomic Profiling: Chromatin immunoprecipitation sequencing (ChIP-seq) for histone modifications and transcription factor binding, assay for transposase-accessible chromatin sequencing (ATAC-seq), and chromosome conformation capture techniques [8].
- Transcriptomic Analysis: Single-nucleus RNA sequencing reveals cell-type specific expression patterns in archived specimens [3] [11].
- Disease Mechanism Elucidation: Identification of cell-type specific enhancer elements and their relationship to genetic risk variants for Alzheimer's disease (enriched in microglia) and psychiatric disorders (enriched in neurons) [8].
- Multimodal Analysis: Combined snRNA/T cell receptor sequencing with spatial transcriptomics and whole-genome sequencing from small, frozen clinical specimens [11].
Nuclei isolation from frozen archival brain tissue represents a fundamental advancement in neuroscience research methodology, enabling unprecedented access to the vast resources of biobanked specimens. The superior compatibility of nuclei with frozen tissue, reduced technical artifacts compared to whole-cell approaches, and ability to preserve cellular diversity make this approach indispensable for modern translational neuroscience. By selecting appropriate isolation methodologies and following optimized protocols, researchers can generate high-quality genomic and epigenomic data from archived specimens, accelerating our understanding of neurological and psychiatric disorders.
The integration of single-nucleus RNA sequencing (snRNA-seq) and epigenetics represents a transformative approach in translational research, particularly for investigating complex tissues like the brain. The foundation of this research relies on robust methods for isolating nuclei from frozen postmortem brain tissue, which opens access to invaluable biobanks and rare disease samples that were previously difficult to study [3] [12]. This capability is crucial for advancing our understanding of neurological disorders, cancer heterogeneity, and developmental processes at single-cell resolution.
snRNA-seq overcomes a fundamental limitation of traditional single-cell RNA sequencing by eliminating the need for intact cell dissociation, a process that often compromises neuronal integrity or activates glial cells, leading to artifacts in the data [3]. Furthermore, the use of nuclei rather than whole cells enables the transcriptomic profiling of frozen tissues, including postmortem samples from individuals with neurodegenerative conditions such as Alzheimer's disease, thereby providing unprecedented access to the cellular and molecular underpinnings of human disease [3].
Core Protocol: Nuclei Isolation from Frozen Postmortem Brain Tissue
The quality of nuclei isolation directly influences all downstream molecular analyses and biological interpretations. This section details optimized methodologies validated on challenging frozen brain tissues.
Optimized Isolation Protocol for Frozen Brain Tissue
An optimized protocol for frozen pediatric glioma tissue demonstrates that a simplified, rapid approach can yield high-quality nuclei. This method balances purity, yield, and structural integrity, which is essential for successful snRNA-seq [12].
Key Steps:
- Tissue Preparation: Cut 20–50 mg of frozen tissue on dry ice or in ice-cold lysis buffer.
- Homogenization: Dounce the tissue sample to open cell walls while preserving nuclear integrity.
- Filtration: Pass the homogenate through a cell strainer to remove large debris.
- Washing: Perform 2–3 washes with lysis buffer without detergent to remove cellular debris and ambient RNA without permeabilizing nuclear walls [12].
This protocol is fast (under 30 minutes), cost-effective, and results in intact nuclei with minimal debris, as evidenced by very low proportions of mitochondrial reads (typically under 1%) in subsequent sequencing data [12]. The number of washes can be adjusted based on starting material; two washes may be preferable for very low-input samples to minimize nucleus loss [12].
Comparative Analysis of Isolation Methods
Different nuclei isolation strategies can significantly impact experimental outcomes. A systematic comparison of three mechanistically distinct methods highlights protocol-dependent variations in yield, integrity, and downstream data quality [3].
Table 1: Comparison of Nuclei Isolation Methods for Brain Tissue
| Method | Key Principle | Nuclei Yield | Nuclei Integrity | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Sucrose Gradient Centrifugation [3] | Manual homogenization followed by sucrose gradient ultracentrifugation | ~60,000 nuclei per mg input [3] | ~85% intact nuclei [3] | Well-established, cost-effective | Person-to-person variability, requires ultracentrifuge |
| Spin Column-Based Method [3] | Filtration and purification through specialized columns | 25% lower than other methods [3] | ~35% intact nuclei [3] | Faster processing, no specialized machinery | Notable aggregation and debris, requires specific columns |
| Machine-Assisted Platform [3] | Automated, standardized homogenization and isolation | ~60,000 nuclei per mg input [3] | ~100% intact nuclei [3] | High throughput, minimal variability, negligible debris | Requires specialized, costly equipment and cartridges |
The following workflow diagram summarizes the optimized protocol for isolating nuclei from frozen brain tissue:
Advanced Applications: Integrating snRNA-seq and Epigenetics in Translational Research
The synergy between snRNA-seq and epigenetic analysis provides a powerful framework for addressing complex biological questions in translational neuroscience.
Cell Type-Specific Epigenomics with FANS
For epigenetic studies, isolating specific cell types is often necessary. Fluorescent-Activated Nuclei Sorting (FANS) enables the purification of neuronal nuclei from heterogeneous suspensions for downstream bulk or single-cell epigenomic analyses [2].
Immunostaining for FANS:
- Primary Antibody Incubation: Stain nuclei with an antibody for a neuronal marker (e.g., NeuN) for 30 minutes on ice [2].
- Washing: Pellet nuclei by centrifugation (5 min at 400 rcf) to remove unbound antibody [2].
- Secondary Antibody Incubation: Add a fluorescently-labeled secondary antibody and incubate for 15 minutes in the dark on ice [2].
- Final Washes: Perform two washes to remove unbound secondary antibody [2].
This protocol has been successfully used to isolate neuronal nuclei from chimpanzee cerebral cortex for methylome sequencing, achieving sufficient coverage for genome-wide epigenetic analysis [2]. The process is visualized below:
Key Translational Applications
The application of these techniques spans multiple domains of biomedical research:
- Decoding Brain Complexity: snRNA-seq enables detailed mapping of neuronal and glial diversity in the human brain, providing insights into cortical development, neurogenesis, and the cellular mechanisms underlying disorders like autism and intellectual disabilities [13].
- Advancing Cancer Research: In pediatric gliomas and other brain tumors, snRNA-seq reveals tumor heterogeneity, identifies rare cell populations driving progression, and uncovers molecular signatures of treatment resistance [13] [12]. This granular view is invaluable for developing personalized treatment strategies.
- Elucidating Epigenetic Mechanisms in Disease: Integration of snRNA-seq with epigenetic technologies allows researchers to investigate mechanisms like DNA methylation and histone modifications in specific brain cell types. This is crucial for understanding their roles in neurodegenerative diseases, aging, and synaptic plasticity [14] [2].
Quality Control and Data Analysis
Rigorous quality control is essential for ensuring data integrity and accurate biological interpretation.
Computational Quality Control with QClus
Quality Clustering (QClus) is a computational approach that improves snRNA-seq data quality by removing empty and highly contaminated droplets using multiple contamination metrics [15]. This preprocessing step is particularly valuable for samples with high levels of ambient RNA contamination, a common challenge in complex tissues like the brain [15].
Impact of Isolation Methods on Data Quality
The choice of nuclei isolation protocol directly influences downstream transcriptional profiles and cell type detection:
- Cell Type Proportions: Different isolation methods capture varying proportions of cell types. For example, sucrose gradient centrifugation captured the largest proportion of astrocytes (13.9%), while a machine-assisted platform recovered the most microglia (5.6%) and oligodendrocytes (15.9%) [3].
- Transcriptional Homogeneity: Isolation workflows differentially affect the transcriptional consistency within cell populations, as measured by metrics like the ratio of global unshifted entropy (ROGUE) [3].
- Contamination Levels: Protocols vary in their effectiveness at minimizing contamination from ambient, mitochondrial, and ribosomal RNAs, all of which can overwhelm sequencing data and reduce detection of informative transcripts [3].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these advanced protocols requires specific reagents and tools. The following table details key solutions for nuclei isolation and downstream applications.
Table 2: Essential Research Reagents for Nuclei Isolation and Downstream Applications
| Reagent/Tool | Specific Function | Application Examples |
|---|---|---|
| 10X Genomics Chromium Nuclei Isolation Kit [2] | Standardized nuclei isolation with RNase inhibition | Nuclei preparation for single-cell sequencing from small (25 mg) frozen tissue inputs [2] |
| Dounce Homogenizer [12] | Mechanical tissue disruption while preserving nuclear integrity | Critical step in optimized protocol for frozen brain tissue [12] |
| Sucrose Gradient Solutions [3] | Density-based purification of nuclei from debris | Centrifugation-based isolation method for brain tissue [3] |
| Anti-NeuN Antibody [2] | Immunostaining marker for neuronal nuclei | Fluorescent-activated nuclei sorting (FANS) to enrich neuronal populations [2] |
| Propidium Iodide (PI) / Acridine Orange (AO) [2] | Nuclear viability staining for quantification | Distinguishing intact nuclei from debris during counting [2] |
| QClus Algorithm [15] | Computational filtering of contaminated droplets | Preprocessing snRNA-seq data to remove ambient RNA contamination [15] |
The integration of robust nuclei isolation methods with snRNA-seq and epigenetic technologies represents a powerful framework for advancing translational research on frozen postmortem brain tissues. The optimized protocols detailed here provide a clear pathway for researchers to leverage precious archival samples, from rare neurological disorders to unique non-human primate models. As these methods continue to evolve and become more accessible, they will undoubtedly accelerate the pace of discovery in brain research, cancer biology, and the development of targeted therapeutics. The critical importance of method selection and quality control at every stage cannot be overstated, as these factors directly shape data quality, biological interpretation, and ultimately, the translational impact of the research.
Navigating the initial stages of experimental design is paramount for the success of single-nucleus RNA sequencing (snRNA-seq) from frozen postmortem brain tissue. The quality of your data is intrinsically linked to the quality of your starting material and the choices made before the first step of nuclei isolation. This application note details the critical pre-analytical variables—tissue quality and sample limitations—that researchers must evaluate to ensure robust, reproducible, and biologically relevant outcomes in their studies of neurological development, aging, and disease.
Assessing Tissue Quality and Pre-processing
The integrity of frozen postmortem brain tissue is a primary determinant of nuclei isolation success. Key factors include the postmortem interval (PMI), tissue preservation method, and storage conditions, which collectively influence RNA integrity and nuclear architecture.
Table 1: Key Pre-analytical Variables and Their Impacts on Tissue Quality
| Variable | Consideration | Potential Impact on Nuclei & Data |
|---|---|---|
| Postmortem Interval (PMI) | Time between death and tissue preservation. | Shorter intervals are ideal; longer PMIs can reduce RNA integrity and increase cellular debris [2]. |
| Cryopreservation Method | Rapid freezing vs. slow freezing; use of cryoprotectants. | Rapid freezing between cooled metal plates minimizes ice crystal artifacts that disrupt cellular and nuclear membranes [16]. |
| Storage Duration & Temperature | Long-term storage at -80°C or in liquid nitrogen. | Prolonged storage can lead to freeze-drying and degradation, though snRNA-seq remains viable on long-term stored samples [16] [5]. |
| Tissue Dissection | Macro-dissection or micro-punching of specific brain regions on dry ice. | Accurate dissection of the region of interest is critical; small punches (~25 mg) are feasible but require protocol miniaturization [2]. |
A major challenge specific to primate brains is their high proportion of white matter, which leads to elevated levels of myelin debris during homogenization [2]. Furthermore, the inherent RNA degradation in postmortem samples must be acknowledged. While RNA Integrity Number (RIN) is a common metric, for snRNA-seq, the focus shifts to nuclear RNA, which is generally more stable. However, reduced RNA integrity can still compromise cDNA synthesis and library complexity [2].
Navigating Sample Amount and Cell Type Bias
A significant limitation in brain research, especially with rare specimens or small brain regions, is the low amount of starting tissue. Furthermore, the isolation process itself can inadvertently skew the cellular representation in the final data.
Table 2: Addressing Sample Limitations and Technical Biases
| Challenge | Consideration | Recommended Mitigation Strategy |
|---|---|---|
| Low Input Material | Small biopsies or micro-dissected regions (10-50 mg). | Optimized lysis and wash steps can yield sufficient nuclei from as little as 15-25 mg of tissue, avoiding ultra-centrifugation to maximize recovery [2] [5] [17]. |
| Cell Type Isolation Bias | Certain neuronal subtypes or fragile cells may be lost or underrepresented during homogenization. | The choice of nuclei isolation protocol directly influences the proportions of captured cell types [3]. Validation (e.g., with NeuN staining) is crucial [2]. |
| Ambient RNA Contamination | RNA released from lysed cells can adhere to intact nuclei, creating background noise. | Isolation methods differentially influence ambient RNA levels. Computational tools (e.g., SoupX) can post-process data, but high-quality isolation is the best defense [3] [5]. |
The isolation technique itself is a major experimental variable. Comparative studies show that different methods—sucrose gradient centrifugation, spin column-based, and machine-assisted platforms—yield nuclei with different integrity, purity, and subsequently, capture different cell type proportions in snRNA-seq [3]. For instance, one study found that a column-based method yielded only 35% intact nuclei, whereas a machine-assisted method preserved nearly 100% [3]. This underscores the need to select a protocol aligned with your specific research goals.
Essential Reagent Solutions
The following reagents are critical for maintaining nuclear integrity and RNA quality throughout the isolation process.
Table 3: Research Reagent Solutions for Nuclei Isolation
| Reagent | Function | Application Notes |
|---|---|---|
| RNase Inhibitor | Prevents degradation of nuclear RNA. | Essential in all buffers. Concentration is critical (e.g., 0.2 U/μL) [17]. |
| Detergent (e.g., NP-40, Triton X-100) | Disrupts cellular membranes to release nuclei. | Concentration is critical (e.g., 0.05% NP-40); optimal concentration lyses cells without damaging nuclei [5] [17]. |
| Bovine Serum Albumin (BSA) | Reduces non-specific binding and nuclei loss. | Added to wash buffers (e.g., 2-5%) and used to pre-coat tips and tubes to improve nuclei recovery [5] [17]. |
| Dounce Homogenizer | Provides controlled mechanical dissociation. | Pestle clearance (loose vs. tight) and number of strokes must be optimized for brain tissue [5]. |
| Iodixanol Density Gradient | Purifies nuclei by removing cellular debris and myelin. | Effective for cleaning nuclei preparations from lipid-rich brain tissue [5]. |
| Cryoprotectants (e.g., Sucrose, Ethylene Glycol) | Reduce ice crystal formation during tissue freezing. | Protocols for light microscopy or immunofluorescence use different cryoprotectant recipes for optimal structural preservation [18]. |
Experimental Workflow: From Tissue to Analysis
The journey from frozen tissue block to high-quality data involves a series of deliberate steps, each with specific quality control checkpoints. The following diagram outlines the core workflow and critical decision points.
Diagram: Core workflow for nuclei isolation from frozen brain tissue, highlighting critical quality control (QC) checkpoints.
Key Recommendations for Experimental Design
- Prioritize Tissue Characterization: Secure detailed metadata for each sample, including PMI, preservation method, and storage history. This information is indispensable for interpreting data variability and batch effects.
- Pilot Studies are Crucial: Before processing precious samples, conduct pilot experiments with spare tissue to optimize homogenization intensity (Dounce strokes) and lysis time for your specific tissue type and equipment.
- Validate Cell Type Recovery: Use immunohistochemistry or flow cytometry with markers like NeuN to confirm that your isolation protocol does not disproportionately lose specific cell populations, such as neurons or microglia [2] [3].
- Plan for Downstream Applications: The intended snRNA-seq protocol (e.g., 10X 3' RNA-seq vs. ATAC-seq) and any desired bulk sequencing from sorted nuclei will influence the required nuclei quality, quantity, and handling [2] [5]. For example, enzymatic methyl-seq is recommended for methylome profiling from low-input DNA derived from sorted nuclei [2].
By thoroughly addressing these considerations before beginning wet-lab work, researchers can significantly enhance the reliability and impact of their single-nucleus genomic studies on the frozen postmortem brain.
Step-by-Step: Optimized Protocols for Nuclei Isolation and FANS
The isolation of high-quality nuclei from frozen postmortem brain tissue is a critical first step for single-nucleus genomic and epigenomic analyses. The success of these downstream applications is fundamentally dependent on the careful preparation of reagents that balance effective tissue homogenization with the preservation of nuclear integrity [2]. This protocol details the preparation of specialized lysis buffers, sucrose solutions, and protective additives specifically optimized for the challenges presented by frozen postmortem brain tissue, which often contains high levels of myelin debris and exhibits reduced nucleic acid integrity [2] [12]. The formulations provided herein are designed to support researchers in constructing a robust foundation for nuclei isolation, enabling the study of previously inaccessible tissues from humans and non-human primates.
Key Reagent Formulations
Homogenization and Lysis Buffers
Effective nuclei isolation requires buffers that efficiently lyse cellular membranes while keeping nuclear membranes intact. The table below compares several optimized buffer formulations.
Table 1: Composition of Homogenization and Lysis Buffers for Nuclei Isolation
| Component | 10X Genomics-Based Homogenization Buffer (1X HB+) [19] | Low-Input Tissue Lysis Buffer [5] | Simplified Brain Tumor Protocol Lysis Buffer [12] |
|---|---|---|---|
| Sucrose | 640 mM | - | - |
| NaCl | 20 mM | 10 mM | - |
| Tris-HCl | 20 mM (pH 7.5) | 10 mM (pH 7.4) | - |
| MgCl₂ | 6 mM | 3 mM | - |
| Detergent | 0.05% Igepal, 0.0025% Digitonin | 0.05% NP-40 | Not Specified |
| Stabilizers | 0.08% BSA, 2 mM DTT | 5% BSA, 0.25% Glycerol | - |
| Inhibitors | 2X Protease Inhibitor | 40 U/mL RNase Inhibitor | - |
Preparation Notes:
- The 10X Genomics-Based Buffer is prepared as a 2X stock solution (2X HB-) first, which is then diluted to 1X and supplemented with detergents to create the final homogenization buffer (1X HB+) [19].
- The Low-Input Tissue Lysis Buffer is noted for its versatility across different cancer tissues, including brain, and is used ice-cold [5].
- All reagents should be prepared with nuclease-free water, and stocks should be aliquoted and stored at recommended temperatures to maintain efficacy.
Sucrose and Density Gradient Solutions
Sucrose solutions and density gradients are essential for purifying nuclei from cellular debris and myelin.
Table 2: Density Gradient Media for Nuclei Purification
| Solution Type | Composition | Purpose | Reference |
|---|---|---|---|
| Gradient Medium (GM) | 50% Optiprep, 5 mM CaCl₂, 3 mM MgCl₂, 10 mM Tris pH 7.5, 0.04% BSA, 1 mM DTT | Creates a density cushion for nuclei purification during centrifugation. | [19] |
| 29% Optiprep Cushion | 29% Optiprep, Diluted with ODM (150 mM KCl, 30 mM MgCl₂, 60 mM Tris pH 8, 250 mM Sucrose) | Serves as a purification cushion layered underneath the sample. | [19] |
| Sucrose-Containing Buffer | 640 mM Sucrose, 20 mM NaCl, 6 mM MgCl₂, 20 mM Tris pH 7.5, 0.08% BSA | Provides osmotic support and helps maintain nuclear integrity in homogenization buffers. | [19] |
Preparation Notes:
- Optiprep-based solutions are preferred for their ability to form stable gradients that effectively separate intact nuclei from lighter debris [19] [5].
- The 29% Cushion must be prepared precisely and layered carefully to avoid mixing with the sample suspension [19].
Protective Additives and Stabilizers
Protective additives are crucial for maintaining nucleic acid integrity and preventing enzymatic degradation during the isolation process.
Table 3: Protective Additives for Nuclei Isolation Buffers
| Additive Category | Specific Examples | Concentration | Function |
|---|---|---|---|
| RNase Inhibitors | Protector RNase Inhibitor | 40 U/mL [5] | Prevents degradation of nuclear RNA. |
| Protease Inhibitors | Commercial Protease Inhibitor Cocktail | 2X [19] | Prevents protein degradation. |
| Reducing Agents | Dithiothreitol (DTT) | 1-2 mM [19] | Maintains a reducing environment, stabilizes proteins. |
| Stabilizing Proteins | Bovine Serum Albumin (BSA) | 0.04% - 1% [19] [5] | Stabilizes nuclear membranes, reduces sticking. |
| Osmotic Stabilizers | Glycerol | 0.25% [5] | Provides osmotic support and stabilization. |
Experimental Protocols for Reagent Application
Protocol 1: Nuclei Isolation from Frozen Brain Tissue Using a Sucrose-Optiprep Gradient
This protocol, adapted for frozen postmortem brain tissue, utilizes the sucrose and Optiprep solutions detailed in Section 2.1 and 2.2 [19].
Workflow Overview:
Detailed Steps:
- Homogenization: On dry ice, cut ~25-50 mg of frozen cerebral cortex tissue. Immediately transfer the tissue to a pre-chilled homogenizer containing 1 mL of ice-cold Homogenization Buffer (1X HB+, see Table 1). Homogenize with 10 gentle manual strokes using a loose pestle (Pestle A), followed by 10 strokes with a tight pestle (Pestle B) [19] [2].
- Filtration and Initial Spin: Pass the homogenate through a 70 µm cell strainer. Wash the homogenizer and strainer with 1 mL of HB- buffer (Homogenization Buffer without detergent). Transfer the filtered lysate to a tube and centrifuge at 500g for 5 minutes in a swinging bucket rotor. Carefully remove the supernatant [19].
- Density Gradient Purification: Resuspend the pellet in HB- buffer to a final volume of 520 µL. Add an equal volume (520 µL) of Gradient Medium (GM, see Table 2) and mix gently. In a new tube, layer 770 µL of a 29% Optiprep Cushion. Carefully layer the nuclei-GM mixture on top of the cushion without disturbing it.
- Centrifugation and Collection: Centrifuge the layered tubes at a minimum of 3,000 rcf (up to 9,000 rcf) in a swinging bucket rotor at 4°C for 20 minutes with the brake disengaged. After centrifugation, carefully aspirate the supernatant. Resuspend the purified nuclear pellet in an appropriate resuspension buffer (e.g., 1X Nuclei Buffer with 1% BSA) for counting and downstream applications [19].
Protocol 2: Simplified Isolation for Low-Input Frozen Tissue
This protocol is optimized for low-input (15-50 mg) cryopreserved tissues, minimizing steps and reagent costs while maintaining yield [12] [5].
Workflow Overview:
Detailed Steps:
- Tissue Mincing and Lysis: On dry ice, mince 15-50 mg of frozen tissue into small pieces using a scalpel in a pre-cooled mortar. Transfer the tissue to a tube containing 3 mL of ice-cold Lysis Buffer (see Table 1) [5].
- Homogenization: Use a Dounce homogenizer. The number of strokes and the pestle type (loose or tight) should be optimized for the specific tissue type. For brain tissue, start with 10-15 strokes with a loose pestle.
- Filtration and Washing: Incubate the homogenate on ice for 5 minutes. Stop the lysis by adding 5 mL of ice-cold Nuclei Washing Buffer. Filter the suspension through a 30 µm strainer. Centrifuge at 1000g for 10 minutes at 4°C. For purification, wash the pellet 2-3 times by resuspending in 1 mL of Wash Buffer and centrifuging. Two washes may be preferred for very low-input samples to minimize nuclei loss [12].
- Optional FACS Sorting: For the highest purity, resuspend the nuclei in a staining buffer containing a viability dye like 7-AAD or DAPI. Sort the nuclei using a flow sorter to collect intact, positive events, which significantly reduces background debris [5].
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for Nuclei Isolation
| Reagent Solution | Function | Key Considerations |
|---|---|---|
| Homogenization Buffer (with Sucrose) | Provides osmotic stability and initial cell membrane lysis. | The concentration of sucrose (e.g., 640 mM) is critical for maintaining nuclear integrity during homogenization [19]. |
| Detergent Solution (Igepal/Digitinin) | Completes cell lysis and permeabilizes nuclear membranes for antibody staining. | Concentration must be carefully optimized; too high can damage nuclei, too low reduces lysis efficiency [19]. |
| Density Gradient Medium (Optiprep) | Purifies nuclei by separating them from lighter cellular debris and myelin. | Essential for postmortem brain tissue with high debris [2] [19]. |
| RNase/Protease Inhibitor Cocktails | Preserves nucleic acids and nuclear proteins from enzymatic degradation. | Critical for postmortem tissues with potentially reduced integrity; must be added fresh [2] [5]. |
| Nuclei Resuspension Buffer (with BSA) | Stabilizes isolated nuclei for short-term storage and downstream assays. | BSA (e.g., 0.04-1%) prevents nuclei from adhering to tube walls [19] [5]. |
| Fluorescent Stains (DAPI, 7-AAD) | Enables quantification and sorting of nuclei by flow cytometry (FANS). | Allows for gating on DAPI-positive/7-AAD-positive events to select intact nuclei for sequencing [2] [5]. |
The precise preparation of lysis buffers, sucrose solutions, and protective additives forms the cornerstone of successful nuclei isolation from challenging frozen postmortem brain samples. The protocols and formulations provided here address specific challenges such as high myelin content and low starting material. By adhering to these detailed reagent preparation guidelines, researchers can ensure the isolation of high-quality nuclei suitable for advanced single-nucleus genomic and epigenomic studies, thereby maximizing the scientific return from precious and irreplaceable tissue resources.
The choice of homogenization technique is a critical determinant of success in single-nucleus RNA sequencing (snRNA-seq) studies using frozen postmortem brain tissue. This protocol evaluation demonstrates that Dounce homogenization remains the gold standard for its balance of yield and preservation of nuclear RNA integrity, while mechanical techniques offer advantages in throughput and consistency for specific applications. The optimal method varies significantly across different brain regions due to their unique cellular compositions and structural characteristics, with methodological decisions directly impacting cell type representation, RNA quality, and subsequent data interpretation in neurodegenerative disease research.
Effective homogenization is the foundational step in nuclei isolation from frozen postmortem brain tissue, requiring complete tissue disruption while maintaining nuclear membrane integrity. Single-nucleus RNA sequencing (snRNA-seq) has become indispensable for investigating cellular heterogeneity in complex tissues like the brain, particularly because it enables the utilization of frozen tissues and difficult-to-isolate cell types [3]. For postmortem brain research, where fresh tissue dissociation is often impractical, nuclei isolation from frozen specimens provides unique access to the transcriptomic landscape of the human brain in both health and disease states.
The mechanical forces applied during homogenization must be carefully balanced to liberate nuclei from the dense extracellular matrix of various brain regions without causing nuclear rupture or RNA degradation. This balance is particularly crucial when working with frozen tissues, where ice crystal formation can already compromise structural integrity. The choice between gentle Dounce homogenization and more vigorous mechanical methods must be carefully considered in the context of specific brain region characteristics and research objectives.
Comparative Analysis of Homogenization Techniques
Dounce Homogenization
Dounce homogenization utilizes a manually operated glass pestle to apply controlled shear forces through a grinding and twisting motion. This method provides real-time tactile feedback, allowing experienced technicians to adjust pressure and stroke count based on tissue resistance.
- Mechanism: Glass-on-glass shearing with loose (clearance ~0.003-0.006 inches) and tight (clearance ~0.001-0.003 inches) pestles in sequence [20] [21]
- Optimal Applications: Cortical regions, hippocampus, and other architecturally complex regions requiring precise control
- Key Advantages: Adjustable intensity, minimal heat generation, compatibility with diverse buffer systems, preservation of nuclear integrity
- Documented Performance: When applied to mouse brain cortex, Dounce homogenization in lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.01875% NP-40) yielded approximately 2 million nuclei from 30mg of tissue, with 85% structural integrity retention [3]
Mechanical Homogenization
Mechanical methods encompass various automated systems including blade homogenizers, bullet blenders, and rotor-stator systems that standardize disruption through controlled mechanical energy.
- Mechanism: High-speed blending with stainless steel beads (Bullet Blender) or rotating blades [22]
- Optimal Applications: White matter-rich regions, myelinated structures, and high-throughput studies
- Key Advantages: Reproducibility, reduced operator variability, faster processing times, handling of tougher tissues
- Documented Performance: The Bullet Blender system (setting 4 for 4 minutes with 3.2mm stainless steel beads) effectively homogenized frozen mouse and human brain tissues when combined with Nuclei EZ lysis buffer, though nuclear integrity was slightly reduced compared to optimal Dounce homogenization [22]
Table 1: Quantitative Comparison of Homogenization Techniques for Brain Regions
| Parameter | Dounce Homogenization | Mechanical Homogenization |
|---|---|---|
| Nuclei Yield (per mg cortex) | ~60,000 nuclei/mg [3] | Comparable yield, method-dependent [3] |
| Nuclear Integrity | 85% intact nuclei [3] | Varies (35%-100%) by system [3] |
| Processing Time | ~30-45 minutes (including setup) | 5-15 minutes (hands-on time) |
| Inter-operator Variability | Higher (technique-dependent) [3] | Lower (standardized settings) [3] |
| Debris Generation | Low to moderate [21] | Moderate to high (system-dependent) |
| Cell Type Bias | Better neuronal representation [3] | Potential glial enrichment [3] |
Table 2: Brain Region-Specific Technical Considerations
| Brain Region | Structural Challenges | Recommended Technique | Protocol Modifications |
|---|---|---|---|
| Prefrontal Cortex | Complex laminar architecture | Dounce homogenization | Extended incubation (2-5 min) post-homogenization |
| Cerebellum | Dense granular layer | Mechanical homogenization | Reduced bead size (0.5-1mm), shorter duration |
| White Matter Tracts | High myelin content | Mechanical + density gradient | Additional myelin removal steps [23] |
| Brainstem | Mixed neuronal/fiber composition | Modified Dounce | Increased buffer volume (2:1 v/w) |
| Hippocampus | Delicate pyramidal neurons | Gentle Dounce | Reduced stroke count (10-15 tight pestle) |
Detailed Experimental Protocols
Dounce Homogenization Protocol for Frozen Postmortem Cortex
Reagents and Equipment:
- Nuclei Lysis Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.01875% NP-40, 0.2 U/μL RNase inhibitor) [21]
- Nuclei Wash Buffer (2% BSA in 1x PBS, 0.2 U/μL RNase inhibitor)
- Dounce homogenizer (2mL or 7mL based on sample size)
- Pre-chilled 70μm cell strainers
- Refrigerated centrifuge with swinging bucket rotor
Stepwise Procedure:
- Tissue Preparation: Thaw 20-30mg frozen cortical tissue on ice for 10 minutes in 1mL ice-cold lysis buffer [21].
- Initial Homogenization: Transfer tissue to pre-chilled Dounce on ice. Perform 15 strokes with loose pestle at ~0.5 seconds per stroke with 180° rotation.
- Secondary Homogenization: Continue with tight pestle for 30 strokes, pausing every 10 strokes for 30-second rest periods on ice.
- Incubation: Let homogenate rest on ice for 2 minutes to complete lysis [21].
- Filtration: Strain through 70μm filter into BSA-blocked 15mL tube.
- Wash: Add 4mL wash buffer through filter, invert 5 times to mix.
- Centrifugation: Spin at 500×g for 10 minutes at 4°C [21].
- Resuspension: Carefully resuspend pellet in 1mL wash buffer using wide-bore pipette tips.
Critical Steps:
- Maintain consistent stroke rhythm and rotation
- Monitor lysis progress visually - homogeneous milky suspension indicates complete disruption
- Avoid bubble formation during homogenization
- Work rapidly to minimize RNase activity
Mechanical Homogenization Protocol for Myelinated Brain Regions
Reagents and Equipment:
- Nuclei EZ Lysis Buffer (Sigma Nuc101) with RNase inhibitor (0.04U/μL)
- Stainless steel beads (3.2mm diameter)
- Bullet Blender homogenizer (Next Advance) or equivalent
- Subsequent purification reagents (iodixanol or sucrose gradients)
Stepwise Procedure:
- Tube Assembly: Place 20-50mg frozen tissue in 1.5mL tube with 1mL lysis buffer and single stainless steel bead.
- Homogenization: Process at speed setting 4 for 4 minutes at 4°C [22].
- Intermediate Collection: Briefly centrifuge tubes to collect aerosol and remove beads.
- Filtration: Pass homogenate through 70μm then 35μm filters sequentially.
- Centrifugation: Spin at 700×g for 5 minutes at 4°C.
- Wash: Resuspend in fresh lysis buffer, incubate 2 minutes on ice, and recentrifuge.
- Purification: Proceed to density gradient separation for myelin removal.
Critical Steps:
- Pre-cool blender chamber and samples
- Optimize time/speed settings for specific brain region
- Include appropriate debris removal steps
- Process control samples simultaneously to ensure consistency
The Scientist's Toolkit: Essential Research Reagents
Table 3: Critical Reagents for Nuclear Isolation from Frozen Brain Tissue
| Reagent/Category | Specific Examples | Function & Importance |
|---|---|---|
| Detergents | NP-40, Tween-20 | Selective plasma membrane disruption while preserving nuclear envelope integrity [24] [21] |
| RNase Inhibitors | Protector RNase Inhibitor, RiboLock | Preserve nuclear RNA integrity during processing [24] |
| Buffer Components | Tris-HCl, NaCl, MgCl₂, DTT | Maintain osmotic balance and nuclear stability [24] |
| Density Gradient Media | Iodixanol, Sucrose, Percoll | Debris removal and nuclei purification [6] [22] |
| Protease Inhibitors | Complete Protease Inhibitor Cocktail | Prevent nuclear protein degradation [6] |
| Blocking Agents | Bovine Serum Albumin (BSA) | Reduce non-specific binding and nuclear clumping [20] [24] |
Technical Considerations for Brain Region Applications
Region-Specific Optimization Strategies
Different brain regions present unique challenges that necessitate methodological adaptations:
Prefrontal Cortex: This architecturally complex region with well-defined cortical layers benefits from gentle Dounce homogenization with extended incubation (2-5 minutes) post-homogenization to ensure complete liberation of nuclei from the dense neuropil without excessive shear forces [3].
Cerebellum: The high cellular density of the granular layer requires more vigorous disruption. Mechanical homogenization with smaller beads (0.5-1mm) for shorter durations provides more consistent yields while minimizing nuclear damage [21].
White Matter Tracts: Regions with high myelin content such as the corpus callosum necessitate combined mechanical homogenization and density gradient separation. The addition of specific myelin removal beads or iodixanol gradient centrifugation effectively reduces contaminating myelin debris that can interfere with downstream applications [23].
Brainstem: The heterogeneous composition of neuronal populations and fiber tracts in brainstem nuclei benefits from modified Dounce homogenization with increased buffer volume (2:1 volume-to-weight ratio) to ensure complete tissue dispersion.
Quality Assessment and Troubleshooting
Nuclear Quality Metrics:
- Structural Integrity: ≥90% single, round nuclei with sharp borders under microscopy [25]
- Viability: Trypan Blue exclusion with ≤10% nuclear staining
- RNA Integrity: RIN >7.0 for postmortem samples (when assessing bulk RNA)
- Yield Validation: Consistency with expected yields (approximately 60,000 nuclei/mg cortical tissue) [3]
Common Issues and Solutions:
- Low Yield: Increase homogenization intensity or duration; verify buffer composition
- Poor Integrity: Reduce mechanical force; shorten processing time; increase RNase inhibition
- Excessive Debris: Incorporate density gradient purification; optimize filtration strategy
- Cell Type Bias: Adjust homogenization method to preserve fragile populations
The selection between Dounce and mechanical homogenization techniques must be guided by both the specific brain region under investigation and the research objectives. Dounce homogenization provides superior preservation of nuclear architecture and neuronal representation, making it ideal for transcriptomic studies of cortical regions where maintaining cellular diversity is paramount. Mechanical methods offer reproducibility and efficiency for processing multiple samples or tougher brain regions with high myelin content.
For research focused on comprehensive cell atlas generation from cortical regions, the protocol should prioritize Dounce homogenization with careful quality control. For studies targeting specific glial populations or working with white matter-rich regions, mechanical homogenization combined with appropriate purification strategies may yield more consistent results. Critically, regardless of the chosen method, rigorous quality assessment and protocol validation using the specific brain region of interest are essential for generating reliable snRNA-seq data from frozen postmortem brain tissue.
The isolation of high-quality nuclei from frozen postmortem brain tissue is a critical step for single-cell and single-nucleus multi-omic studies, including single nucleus RNA sequencing (snRNA-seq) and single nucleus Assay for Transposase-Accessible Chromatin with sequencing (snATAC-seq). Such studies are fundamental to understanding cell-type-specific gene regulatory landscapes in neurodevelopment, aging, and neurodegenerative diseases [2]. However, postmortem brain tissue from non-human primates (NHPs) and humans presents unique challenges, including high levels of myelin debris, reduced RNA integrity, and the frequent unavailability of fresh tissue [2]. This application note details optimized protocols for nuclei purification via ultracentrifugation and density gradient methods, framed within the context of a broader thesis on nuclei isolation from frozen postmortem brain tissue. These protocols are designed to help researchers overcome these challenges to obtain high-purity, high-quality nuclei suitable for downstream genomic analyses.
Key Research Reagent Solutions
The following reagents and kits are essential for successful nuclei isolation and purification.
Table 1: Essential Reagents for Nuclei Isolation and Purification
| Reagent/Kits | Primary Function | Examples & Citations |
|---|---|---|
| Nuclei Isolation Kit | Provides optimized buffers for tissue lysis and nuclei stabilization. | 10X Genomics Chromium Nuclei Isolation Kit [2] |
| Density Gradient Medium | Separates nuclei based on buoyant density, removing debris. | Cesium Chloride (CsCl), Sucrose, Percoll [26] [27] |
| Homogenization Buffer | Lyses cell membranes while preserving nuclear integrity. | Triton X-100, DTT, RNase Inhibitors [2] [17] |
| Fluorescent Stains | Labels nuclei for quantification and sorting. | DAPI, Propidium Iodide (PI), Acridine Orange (AO) [2] [26] |
| Antibodies for Sorting | Enables cell-type-specific nuclei enrichment. | Anti-NeuN for neuronal nuclei [2] |
The performance of different nuclei isolation protocols can be evaluated based on yield, purity, and suitability for sequencing.
Table 2: Quantitative Outcomes of Nuclei Isolation from Challenging Tissues
| Tissue Type / Application | Key Metric | Reported Outcome | Citation |
|---|---|---|---|
| Chimpanzee Cerebral Cortex | Nuclei Yield (from ~25-50 mg) | Sufficient for snRNA-seq/ATAC-seq and methylome sequencing [2] | [2] |
| Chimpanzee Cortex (NeuN+ Sorts) | Post-Sort Validation | Successful 10X 3'-RNA-seq library generation from NeuN+ and NeuN- populations [2] | [2] |
| Plant Leaves (10 species) | Nuclei Quality | High purity and yield suitable for snRNA-seq and Cell Division Cycle analysis [26] | [26] |
| Human Kidney Biopsies | Protocol Duration & QC | Processing in ≤90 min; eliminated debris and avoided stress artifacts [17] | [17] |
| Adenovirus (AdV) DGE-AUC | Sensitivity & Throughput | ~56x sensitivity improvement vs. SV-AUC; 21 samples in 80 min [27] | [27] |
Detailed Experimental Protocols
Optimized Protocol for Frozen Postmortem Primate Brain Tissue
This protocol, adapted for frozen chimpanzee cerebral cortex, is designed for small tissue amounts (~25 mg) and standard lab equipment [2].
Methodology:
- Tissue Dissection: On dry ice, microdissect frozen brain slabs using a biopsy punch to obtain 10-25 mg tissue pieces.
- Nuclei Isolation: Use the 10X Genomics Chromium Nuclei Isolation Kit with critical optimizations:
- Modified Lysis: Precisely optimize the lysis time to prevent under- or over-lysing.
- Enhanced Filtration: Incorporate a filtration step using a Flowmi cell strainer.
- Additional Washes: Include extra wash steps to reduce myelin and debris [2].
- Quantification: Use an automated cell counter (e.g., Countess 3 FL) with viability stains (Acridine Orange & Propidium Iodide). A successful isolation shows a majority of double-stained or PI-positive nuclei [2].
- Immunostaining & Sorting (FANS): For neuronal nuclei enrichment:
- Incubate nuclei with primary antibody (e.g., Anti-NeuN) for 30 minutes on ice.
- Wash, then incubate with a fluorescent secondary antibody for 15 minutes in the dark on ice.
- Perform Fluorescent-Activated Nuclei Sorting (FANS) using a cell sorter (e.g., Sony SH800Z). Use unstained and secondary-antibody-only controls to set gates [2].
- Downstream Applications: Sorted nuclei can be used for snRNA-seq, snATAC-seq, or bulk methylome sequencing. For low-input DNA from sorted nuclei, use kits like the QIAamp DNA Micro Kit. Enzymatic methyl-seq (e.g., NEBNext Enzymatic Methyl-seq Kit) is recommended over bisulfite conversion for better DNA preservation [2].
Density Gradient Ultracentrifugation for Purification
Density Gradient Equilibrium Analytical Ultracentrifugation (DGE-AUC) is an orthogonal method that provides high-resolution separation of nuclei based on buoyant density, effectively removing contaminants [27].
Methodology:
- Gradient Optimization: The core of the method is optimizing the density gradient medium (e.g., Cesium Chloride - CsCl).
- Phase 1 (Screening): Use a broad CsCl density range (e.g., 1.20-1.40 g/mL) at high speed (42 krpm) to identify the approximate buoyant density of your nuclei and the time to equilibrium.
- Phase 2 (Refinement): Test a finer density range (e.g., 1.30-1.35 g/mL) at multiple speeds (e.g., 42 krpm and 25 krpm) to find the density that positions nuclei near the gradient's midpoint for maximal resolution.
- Phase 3 (Finalization): Run a multispeed experiment at the optimized density to select the ideal balance between sensitivity (steeper gradient at higher speed) and resolution (shallower gradient at lower speed) [27].
- Sample Run & Analysis: Load the nuclei suspension mixed with the optimized CsCl density solution into the ultracentrifuge. At equilibrium, nuclei will migrate to their isopycnic position. The separation is analyzed using UV/Vis absorbance and Rayleigh interference [27].
Workflow and Pathway Visualizations
Nuclei Isolation and Purification Workflow from Frozen Brain Tissue
Density Gradient Ultracentrifugation Optimization Strategy
Fluorescence-Activated Nuclei Sorting (FANS) with Anti-NeuN for Neuronal Enrichment
The analysis of cell-type-specific epigenetic alterations in the brain is crucial for understanding development, aging, and neurodegenerative diseases. However, the cellular heterogeneity of brain tissue poses a significant challenge, as traditional genomic methods combine signals across different cell types, obscuring cell-type-specific information [2]. For nonhuman primates and humans, fresh tissue is often inaccessible, making frozen postmortem tissue a critical, though technically challenging, resource [2]. This application note details an optimized protocol for isolating nuclei from frozen postmortem cerebral cortex tissue and enriching for neuronal nuclei using Fluorescence-Activated Nuclei Sorting (FANS) with an antibody against the neuronal nuclei antigen (NeuN). This protocol enables downstream single-cell epigenomic analyses and bulk methylome sequencing from precious and limited sample sources [2].
Background
The NeuN Biomarker
NeuN (Neuronal Nuclei), also known as Fox-3 or Rbfox3, is a neuronal nuclear protein widely used as a biomarker for post-mitotic neurons [28] [29]. Discovered in 1992, it is expressed in the nuclei and, to a lesser extent, the perinuclear cytoplasm of most neurons in the central nervous system of mammals [28]. Its expression begins as neurons mature and becomes a robust indicator of neuronal differentiation [29]. While the vast majority of neurons are NeuN-positive, notable exceptions include Purkinje cells, olfactory mitral cells, and retinal photoreceptors [29]. The protein exists in multiple isoforms, typically observed at 46-48 kDa on Western blots, and functions as a pre-mRNA splicing regulator, playing a role in neuronal gene regulation [30] [29].
Technical Challenges of Postmortem Primate Brain Tissue
Working with frozen postmortem brain tissue, particularly from nonhuman primates, presents specific hurdles that require protocol adaptations [2]:
- High levels of myelin debris can compromise downstream workflows.
- Reduced RNA integrity is common due to postmortem intervals.
- Frequently limited tissue amounts are available, as some brain regions yield small samples.
- Autofluorescence and background fluorescence can reduce signal resolution during flow cytometry.
The protocol outlined below was specifically optimized to address these challenges, incorporating key steps to improve nuclei quality, integrity, and sorting outcomes from small starting amounts (~25 mg) of tissue [2].
Materials and Methods
Research Reagent Solutions
The following table lists essential reagents and equipment required for the successful isolation and sorting of neuronal nuclei.
Table 1: Key Research Reagents and Equipment
| Item | Function/Description | Example Catalog Numbers/Models |
|---|---|---|
| Chromium Nuclei Isolation Kit (10X Genomics) | Core kit for isolating nuclei from small amounts of frozen tissue. | PN-1000494 [2] |
| Anti-NeuN Primary Antibody | Binds to the neuronal marker NeuN (Rbfox3) for immunostaining. | N/A [2] |
| Fluorophore-Conjugated Secondary Antibody | Binds to the primary antibody for fluorescent detection during FANS. | N/A [2] |
| DAPI (4',6-Diamidino-2-Phenylindole) | Membrane-impermeable DNA stain used to identify intact nuclei. | D1306 (Thermo Fisher) [2] [31] |
| Flow Cytometry Cell Sorter | Instrument for analyzing and sorting stained nuclei. | Sony SH800Z Cell Sorter [2] |
| Automated Cell Counter | For accurate quantification of nuclei concentration and viability. | Countess 3 FL (Thermo Fisher) [2] |
| Cell Strainer | Removes large debris and clumps from the nuclei suspension. | Flowmi cell strainer (Bel-Art) [2] |
Nuclei Isolation from Frozen Tissue
This protocol is adapted for ~25-50 mg of frozen postmortem cerebral cortex tissue [2].
- Tissue Dissection: Perform micro-dissection on dry ice using a biopsy punch to obtain the required tissue amount. Weigh the tissue in a pre-chilled microcentrifuge tube.
- Nuclei Lysis and Homogenization: Use the 10X Genomics Chromium Nuclei Isolation Kit with RNase Inhibitor. Optimize the lysis time to balance complete tissue disruption and preservation of nuclei integrity. Mechanically homogenize the tissue on ice.
- Filtration and Washing: Pass the homogenate through a pre-chilled Flowmi cell strainer to remove large debris and myelin. Perform additional wash steps with the kit's buffer to reduce background debris further.
- Nuclei Quantification and Viability:
- Mix 5 µL of nuclei suspension with 5 µL of ReadyCount Red/Green Viability Stain.
- Load onto the Countess 3 FL automated cell counter.
- A successful isolation yields a majority of Propidium Iodide (PI)-positive nuclei, indicating intact, membrane-bound nuclei ready for sorting [2].
Table 2: Quantitative Outcomes of Nuclei Isolation
| Metric | Typical Outcome | Measurement Method |
|---|---|---|
| Starting Tissue Mass | 25 - 50 mg | Laboratory scale [2] |
| Nuclei Integrity | Majority PI-positive | Fluorescent cell counter with PI/AO stain [2] |
| Downstream Methylome Sequencing Coverage | >7X coverage of genome-wide CpGs | Illumina NovaSeq 10X [2] |
| PCR Duplication Rate | 13.8% | Downstream sequencing analysis [2] |
Immunostaining for NeuN
All staining steps should be performed on ice or at 4°C.
- Aliquot Controls: Subdivide the nuclei suspension into separate tubes for an unstained control, a secondary antibody-only control, and the experimental sample.
- Primary Antibody Incubation: Incubate the sample with the anti-NeuN primary antibody for 30 minutes on ice.
- Wash: Pellet nuclei by centrifugation at 400 rcf for 5 minutes. Remove the supernatant to eliminate unbound primary antibody. Resuspend the pellet in a blocking buffer.
- Secondary Antibody Incubation: Add the fluorophore-conjugated secondary antibody to the sample and the secondary-only control. Incubate for 15 minutes in the dark on ice.
- Final Washes: Wash the stained nuclei twice to remove any unbound secondary antibody. Keep samples on ice and protected from light until sorting.
Fluorescence-Activated Nuclei Sorting (FANS)
The workflow for sorting involves sequential gating to isolate a pure population of neuronal nuclei.
Diagram 1: FANS Gating Workflow
- Instrument Setup: Use the unstained control to set appropriate voltages and establish a negative fluorescence gate on the flow cytometer.
- Gating for All Nuclei: Add DAPI to the unstained control and establish a gate for DAPI-positive events, which represent the total population of intact nuclei.
- Background Threshold: Run the secondary antibody-only control with DAPI to set the background threshold for the fluorescence channel detecting the secondary antibody.
- Sorting Neuronal Nuclei: Finally, run the anti-NeuN stained sample with DAPI. Sort the population that is positive for both DAPI and the NeuN signal above the background threshold.
- Collection: Collect the sorted neuronal (NeuN+) nuclei in centrifuge tubes pre-filled with a chilled blocking buffer.
Table 3: Experimental Controls for FANS
| Control Type | Purpose | Key Information Gained |
|---|---|---|
| Unstained Nuclei | To adjust flow cytometer voltages and detect autofluorescence. | Baseline instrument settings [2] |
| Secondary Antibody Only | To account for non-specific binding of the secondary antibody. | Background fluorescence threshold [2] |
| Full Stain (NeuN + Secondary) | The experimental sample for sorting. | Defines the NeuN+ neuronal population [2] |
Downstream Applications
Sorted neuronal nuclei are suitable for a variety of genomic applications:
- Single-Cell Epigenomics: Input sorted or unsorted nuclei suspensions directly into protocols like single-cell ATAC-seq or RNA-seq (e.g., 10X Genomics) [2].
- Bk Methylome Profiling: Use sorted neuronal nuclei for bulk DNA methylation analysis. For the low DNA yields typical from sorted nuclei, use a DNA extraction kit optimized for small inputs (e.g., QIAamp DNA Micro Kit). Enzymatic methyl-seq (e.g., NEBNext Enzymatic Methyl-seq Kit) is recommended over bisulfite conversion for better DNA preservation [2].
Discussion and Considerations
Technical Considerations
- Cell Type Bias: Be aware that some neuronal subtypes do not express NeuN (e.g., Purkinje cells, cerebellar inferior olive neurons), which may introduce a selection bias in the resulting analysis [28] [29].
- Phosphorylation State: The detection of NeuN by common antibodies (like clone A60) can be dependent on the phosphorylation state of the protein. Dephosphorylation may mask the epitope and lead to false-negative results [28].
- Protocol Flexibility: While developed for chimpanzee cortex, the core principles of optimized lysis, filtration, and staining can be adapted for nuclei isolation from other complex frozen tissues, such as mouse placenta or pancreas [31].
Validation
After establishing the protocol, it is critical to validate the sorted populations. This can be achieved by collecting both the NeuN-positive and NeuN-negative fractions and preparing 10X 3'-RNA-seq libraries from each. Sequencing data should show a clear enrichment of neuronal transcripts in the NeuN+ fraction and their depletion in the NeuN- fraction, confirming successful neuronal enrichment [2].
Adapting the Protocol for Low-Input Samples (as low as 15mg)
Single-nuclei RNA sequencing (snRNA-seq) has become an indispensable tool for unraveling cellular heterogeneity in complex tissues, particularly the human brain. While comprehensive cellular atlases have been generated, their translation to clinically relevant samples faces significant hurdles. Research on frozen postmortem brain tissue is often constrained by limited sample availability, as biopsies or rare diseased tissues yield very small quantities. Traditional nuclei isolation protocols typically require substantial starting material (often 30-100 mg), making the study of these precious low-input samples challenging. This application note details an optimized, versatile method for isolating high-quality nuclei from cryopreserved tissues with inputs as low as 15 mg, enabling robust snRNA-seq and unlocking potential for novel discoveries in neurology and drug development.
Optimized Protocol for Low-Input Nuclei Isolation
The following protocol is adapted for minimal tissue input while maintaining high nuclei integrity and yield, specifically tailored for frozen postmortem brain tissue.
Materials and Reagents
Table: Essential Reagents for Nuclei Isolation from Low-Input Samples
| Item | Supplier | Catalogue Number | Purpose |
|---|---|---|---|
| RNase AWAY / RNaseZap | Thermo Fisher Scientific | 10328011 / N/A | Surface decontamination |
| Dounce Homogenizer (with loose & tight pestles) | Fisher Scientific | K885301-0002 / K885303-0007 | Mechanical tissue homogenization |
| 10X PBS, RNase-free | Invitrogen | AM9625 | Buffer component |
| UltraPure BSA (50 mg/mL) | Thermo Fisher Scientific | AM2616 | Reduce nuclei loss, buffer component |
| RNase Inhibitor (e.g., RNasin Plus) | Promega | N2615 | Preserve RNA integrity |
| Triton X-100 or NP-40 | Merck Life Science / Various | T9284-100M / N/A | Cell membrane lysis |
| Iodixanol (Optiprep) | Millipore Sigma | D1556 | Density cushion for purification |
| DAPI (4′,6-diamidino-2-phenylindole) | Sigma-Aldrich | D9542 | Nuclei staining and quantification |
| FlowMi Cell Strainers (40 µM, 70 µM) | Sigma-Aldrich | N/A | Debris filtration |
Step-by-Step Methodology
1. Pre-Homogenization Preparation
- Chill a mortar, pestle, and scalpel on dry ice. Pre-cool a centrifuge to 4°C.
- Pre-chill all buffers on ice. Prepare fresh Lysis Buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl₂, 0.05% NP-40 or Triton X-100) and Nuclei Washing Buffer (0.5X PBS, 5% BSA, 0.25% Glycerol, 40 U/mL RNase inhibitor) [5] [17].
- Decontaminate all work surfaces, equipment, and pipettes with RNase AWAY or RNaseZap to prevent RNA degradation [32].
2. Tissue Mincing and Homogenization
- On dry ice, mince approximately 15 mg of frozen brain tissue into fine pieces using a pre-cooled scalpel in the mortar [5].
- Transfer the minced tissue to a pre-cooled 2 mL tube containing 1 mL of ice-cold Lysis Buffer.
- For brain tissue, homogenize using a Dounce homogenizer. Perform 10-15 strokes with the loose pestle (Pestle A), followed by 10-15 strokes with the tight pestle (Pestle B) [5]. The number of strokes can be optimized for different brain regions or tissue integrity.
- Incubate the homogenate on ice for 5 minutes. Stop the lysis reaction by adding 5 mL of ice-cold Nuclei Washing Buffer [5].
3. Filtration and Purification
- Filter the nuclei suspension sequentially through 70 µM and 40 µM FlowMi cell strainers to remove large debris and clumps [17].
- Centrifuge the filtrate at 600-1000 x g for 5-10 minutes at 4°C. Carefully discard the supernatant [5] [17].
- Resuspend the pellet in 1 mL of Nuclei Washing Buffer. Gently layer this suspension over a 2 mL cushion of 29% iodixanol [5].
- Centrifuge at 1000 x g for 10 minutes at 4°C. Intact nuclei will form a pellet, while debris remains in the iodixanol layer. Discard the supernatant and resuspend the purified nuclei pellet in 100-300 µL of Nuclei Washing Buffer [5].
4. Fluorescence-Activated Nuclei Sorting (FANS)
- To ensure the highest nuclei purity for sequencing, stain the suspension with a viability dye like 7-AAD (10-minute incubation) or DAPI [5] [32].
- Use a fluorescence-activated cell sorter (e.g., BD FACSAria Fusion) with a 70 µm nozzle. Gate on the population of DAPI or 7-AAD-positive events that fall within the expected size range for nuclei (calibrated with size beads) to exclude debris and damaged nuclei [5].
- Collect the sorted nuclei and centrifuge at 1000 x g for 10 minutes at 4°C. Resuspend in an appropriate volume (e.g., 70 µL) of washing buffer for downstream processing [5].
5. Quality Control and Quantification
- Quantify nuclei yield and assess integrity using a hemocytometer or automated cell counter (e.g., Countess II FL) with DAPI staining [17].
- Visually inspect nuclei morphology and confirm the absence of cytoplasmic tags or debris using fluorescence microscopy (e.g., ZEISS Axio Observer 7) [5].
Workflow Visualization
Performance and Validation
This protocol's effectiveness has been validated across multiple cancer tissues, including brain, demonstrating robust performance with low-input samples [5].
Table: snRNA-seq Performance Metrics from Low-Input Samples
| Tissue Type | Input Mass (mg) | Nuclei Yield (after sorting) | Nuclei Profiled per Sample | Key Quality Metrics |
|---|---|---|---|---|
| Brain (and other cancers) | ~15 mg | Not Specified | 1,550 - 7,468 | - nCountRNA ≥ 300- nFeatureRNA: 250-2,500- Mitochondrial % < 10 |
Application to human kidney biopsies preserved in RNAlater also confirms the method's utility for diverse, hard-to-process clinical samples [17]. A comparative study of nuclei isolation methods for brain tissue further underscores that protocol choice significantly impacts yield, purity, and the final transcriptional profile [3].
The Scientist's Toolkit
Table: Key Research Reagent Solutions for Low-Input Nuclei Isolation
| Reagent/Category | Function | Key Consideration for Low-Input Samples |
|---|---|---|
| RNase Inhibitors | Prevents degradation of nuclear RNA | Critical for preserving the limited RNA from small samples. |
| Density Gradient Media (e.g., Iodixanol) | Purifies intact nuclei from cellular debris | Gentle alternative to ultracentrifugation; improves purity without significant yield loss. |
| Fluorescence-Activated Nuclei Sorting (FANS) | Selects intact nuclei based on DNA dye incorporation | Dramatically reduces ambient RNA contamination by removing damaged nuclei; essential for clean data from low-input samples. |
| Anti-Aggregation Buffers (e.g., with BSA) | Prevents clumping of nuclei | Coating tubes and tips with BSA minimizes nuclei loss during handling, maximizing recovery. |
| Dounce Homogenizer | Mechanical tissue disruption | Allows precise, controlled lysis tailored to the tough, lipid-rich environment of brain tissue. |
This detailed protocol provides a reliable framework for isolating high-quality nuclei from low-input, cryopreserved brain samples as small as 15 mg. By integrating optimized mechanical homogenization, rigorous purification via density gradient centrifugation and FANS, and stringent quality control, researchers can effectively leverage precious biobank specimens for high-resolution snRNA-seq studies. This approach empowers the investigation of cellular heterogeneity in neurological diseases and supports the discovery of novel drug targets where tissue quantity is a limiting factor.
Troubleshooting Guide: Solving Common Problems in Brain Nuclei Isolation
Within the broader framework of establishing a robust protocol for nuclei isolation from frozen postmortem brain tissue, the homogenization step is critical. This initial mechanical disruption determines the yield of intact, high-quality nuclei and the level of contaminating debris, thereby directly influencing the success of all downstream genomic applications [3] [5]. For heterogeneous and delicate tissues like the postmortem brain, optimizing the method of homogenization—specifically, the selection of the homogenizer (pestle) and the number of strokes applied—is not a trivial detail but a fundamental requirement for reproducible and high-integrity science [5]. This document provides detailed guidance on optimizing these parameters to overcome the inherent challenges of frozen neural tissue, such as high lipid content and reduced RNA integrity [2].
The Scientist's Toolkit: Essential Homogenization Materials
The following table details key reagents and equipment essential for the homogenization step in nuclei isolation protocols.
Table 1: Key Research Reagent Solutions for Nuclei Isolation Homogenization
| Item | Function & Application | Example References |
|---|---|---|
| Dounce Homogenizer | A glass homogenizer with manually operated pestles used for gentle mechanical tissue disruption. Ideal for freeing nuclei while minimizing damage. | [5] |
| Loose Pestle (Pestle A) | Features a larger clearance (e.g., 0.0025-0.0055 inches). Used for initial, coarse tissue breakdown. | [5] |
| Tight Pestle (Pestle B) | Features a smaller clearance (e.g., 0.0005-0.0025 inches). Used for fine homogenization after the initial coarse step. | [5] |
| Lysis Buffer | A hypotonic buffer containing detergent (e.g., NP-40) to lyse cell membranes and release nuclei while keeping the nuclear membrane intact. | [5] [12] |
| RNase Inhibitor | A critical additive to washing and resuspension buffers that protects RNA from degradation during the isolation process. | [2] [5] |
Pestle Selection: A Guide to Mechanical Disruption
The choice of pestle is a primary variable in balancing yield against nuclear integrity. The Dounce homogenizer, utilized with a series of pestles, is the established instrument for this task, allowing for graded tissue disruption [5].
Pestle Types and Clearances
Two main types of pestles are commonly employed in a sequential manner:
- Loose Pestle (Pestle A): This pestle has a larger clearance, typically between 0.0025–0.0055 inches. Its function is the initial, coarse breakdown of the tissue matrix [5].
- Tight Pestle (Pestle B): This pestle has a much smaller clearance, typically between 0.0005–0.0025 inches. It is used after the coarse homogenization to complete the disruption of individual cells and free the nuclei [5].
The sequential use of loose and tight pestles is a widely recommended strategy for complex tissues like brain and prostate, which require more vigorous homogenization [5]. For tissues with a softer consistency, such as lung, the use of the tight pestle alone may be sufficient, streamlining the process [5].
Stroke Count Optimization by Tissue Type
The number of strokes applied with each pestle must be empirically optimized. Insufficient strokes result in low nuclei yield, while excessive strokes damage nuclear integrity and increase debris.
Table 2: Optimized Homogenization Parameters for Various Cryopreserved Tissues
| Tissue Type | Pestle Sequence | Recommended Stroke Count | Key Observations |
|---|---|---|---|
| Brain (General) | Loose (A) then Tight (B) | Optimized: ~10-15 strokes total | Higher white matter content requires effective homogenization while managing myelin debris [2] [5]. |
| Bladder | Loose (A) then Tight (B) | 10 strokes total | Requires a standardized homogenization force for reproducibility [5]. |
| Lung | Tight (B) only | 10 strokes | Softer tissue consistency allows for a simplified protocol with the tight pestle only [5]. |
| Prostate | Loose (A) then Tight (B) | 10 strokes total | Similar to brain, benefits from sequential pestle use for complete dissociation [5]. |
Protocol-Dependent Variation
It is crucial to note that the optimal stroke count is highly dependent on the specific homogenization buffer and the overall protocol. For instance, a simplified, fast protocol for frozen brain tumor tissue may rely on a specific douncing sequence without specifying an exact stroke count in the available literature, focusing instead on the visual outcome—a debris-free suspension of intact nuclei [12]. Therefore, the values in Table 2 should serve as a starting point for optimization.
Integrated Experimental Protocol for Homogenization
What follows is a detailed methodology for the homogenization of frozen postmortem brain tissue, incorporating the optimization principles for pestle selection and stroke count.
Pre-homogenization Steps
- Tissue Preparation: On dry ice, microdissect the frozen cerebral cortex tissue using a biopsy punch or scalpel. The starting amount should be weighed (~25-50 mg) and transferred to a pre-chilled tube [2].
- Buffer Preparation: Chill an appropriate lysis buffer (e.g., 10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.05% NP-40) on ice [5]. Add RNase inhibitor immediately before use.
Homogenization Procedure
- Initial Suspension: Transfer the frozen tissue pieces to a pre-cooled Dounce homogenizer containing ice-cold lysis buffer.
- Coarse Homogenization: Perform the initial homogenization using the loose pestle (Pestle A). Execute 5-7 steady, controlled strokes, moving the pestle up and down at a rate of approximately one stroke per second. Avoid introducing air bubbles.
- Fine Homogenization: Change to the tight pestle (Pestle B). Execute an additional 5-8 strokes to complete the tissue disruption. The homogenate should appear cloudy and uniform.
- Incubation: Transfer the homogenate to a tube and incubate on ice for 5 minutes to complete cell lysis [5].
- Filtration: Filter the homogenate through a pre-wet 30-40 μm cell strainer to remove large debris and un-dissociated tissue clumps [2] [5]. Proceed with subsequent washing and purification steps as required by the overall nuclei isolation protocol.
Homogenization Workflow and Outcome Relationship
The following diagram illustrates the decision-making process and the downstream consequences of homogenization quality, summarizing the key concepts outlined in this document.
Reducing Debris and Myelin Contamination
In the field of neuroscience research, the isolation of high-quality nuclei from frozen postmortem brain tissue is a critical preparatory step for single-nucleus genomic and epigenomic analyses. However, this process is frequently compromised by two persistent technical challenges: high levels of debris and myelin contamination. These contaminants can severely impact downstream applications by reducing sequencing efficiency, increasing ambient RNA, and obscuring biological signals [2] [3]. The propensity for contamination is particularly pronounced in primate brain tissues, which contain a higher proportion of white matter compared to rodent brains, and in postmortem samples where extended collection intervals and tissue integrity issues further exacerbate these challenges [2]. This application note details optimized methodologies within the broader context of nuclei isolation protocols, specifically addressing debris and myelin reduction to ensure the reliability of subsequent genomic analyses.
Comparative Analysis of Isolation Methods
The selection of an appropriate nuclei isolation strategy fundamentally influences the extent of debris and myelin contamination. Research systematically comparing three distinct isolation approaches—sucrose gradient centrifugation, spin column-based methods, and machine-assisted platforms—reveals significant methodological differences in performance outcomes [3].
Table 1: Performance Comparison of Nuclei Isolation Methods for Brain Tissue
| Isolation Method | Nuclei Yield (per mg tissue) | Intact Nuclei (%) | Debris Level | Myelin Removal |
|---|---|---|---|---|
| Sucrose Gradient Centrifugation | ~60,000 | 85% | Low | Effective [3] |
| Spin Column-Based | ~25% fewer than other methods | 35% | Substantial debris and aggregation | Suboptimal [3] |
| Machine-Assisted Platform | ~60,000 | ~100% | Negligible debris | Effective [3] |
| Commercial Kit (10X Genomics) | Low | Reduced integrity | Elevated debris | Requires optimization [2] [12] |
The data indicates that while sucrose gradient centrifugation and machine-assisted platforms provide superior yield and purity, commercial kits often require significant optimization to achieve comparable results with challenging postmortem primate brain samples [2] [3] [12].
Detailed Protocols for Debris and Myelin Reduction
Optimized Protocol for Frozen Postmortem Primate Brain Tissue
This protocol, adapted for chimpanzee cerebral cortex, addresses specific challenges of nonhuman primate tissue through modified lysis, filtration, and washing procedures [2].
Key Materials:
- Homogenization: Dounce homogenizer with loose (A) and tight (B) pestles.
- Buffers: Lysis buffer without detergent to avoid nuclear permeabilization [12].
- Filtration: 40 μm flowmi cell strainer [2] or similar.
- Centrifugation: Standard laboratory centrifuge (ultracentrifuge not required).
Procedure:
- Tissue Preparation: Microdissect frozen cerebral cortex on dry ice using a 2 mm biopsy punch, yielding 10-25 mg tissue pieces. Weigh 25-50 mg frozen tissue per reaction [2].
- Homogenization: Transfer tissue to a pre-chilled Dounce homogenizer containing ice-cold homogenization buffer. Perform 15 strokes with the 'A' pestle, followed by 15 strokes with the 'B' pestle, keeping the apparatus on ice throughout [33].
- Initial Clarification: Transfer the homogenate to a low-bind tube and centrifuge at 500 × g for 5 minutes at 4°C to pellet nuclei and large debris [33].
- Enhanced Filtration: Carefully aspirate the supernatant and resuspend the pellet in homogenization buffer. Pass the suspension through a 40 μm cell strainer to remove large debris and myelin fragments [2] [12].
- Density Barrier (Optional): For significant myelin contamination, layer the filtered suspension over a 35% iodixanol solution and centrifuge at 10,000 × g for 30 minutes at 4°C with minimal brake. Recover nuclei from the interface between the sample and iodixanol layers [33].
- Washing: If omitting the density barrier, perform 2-3 wash steps by resuspending the nuclei pellet in wash buffer and centrifuging at 500 × g for 5 minutes at 4°C. Two washes are recommended for low-input material to minimize nucleus loss; three washes provide optimal debris removal for adequate starting material [12].
Alternative Strategies for Specific Applications
Partially Automated Protocol: Utilizing a robotic tissue dissociator with pre-cooled nuclei isolation cartridges standardizes homogenization, reducing user variability. For brain tissue, a silica colloid gradient is recommended instead of sucrose for more effective myelin removal [34].
Myelin Removal Density Gradients: Iodixanol (OptiPrep) density gradient centrifugation effectively separates nuclei from less dense myelin fragments. The gradient is prepared by layering the nuclei suspension over a 35% iodixanol solution, which creates a clean interface where purified nuclei collect after centrifugation [33].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Nuclei Isolation and Debris Reduction
| Reagent / Material | Function / Application | Specific Example |
|---|---|---|
| RNase Inhibitor | Prevents RNA degradation during isolation, crucial for transcriptomic integrity [33]. | Added to homogenization and wash buffers (e.g., 0.2 U/μL) [33] [34]. |
| Protease Inhibitor | Preserves nuclear membrane integrity and protein epitopes for immunostaining [33]. | Added to homogenization buffer via tablet dissolution [33]. |
| Iodixanol (OptiPrep) | Medium for density gradient centrifugation; effectively separates nuclei from myelin [33]. | Used to create 35% and 50% solutions for creating a discontinuous gradient [33]. |
| Silica Colloid Solution | Alternative density medium specifically optimized for myelin removal from brain tissue [34]. | Prepared as an 18% solution in Nuclei Storage Reagent [34]. |
| NeuN (Neurological Nuclear) Antibody | Enables fluorescent-activated nuclei sorting (FANS) of neuronal nuclei, reducing non-neuronal contamination [2]. | Used for immunostaining followed by flow sorting to isolate neuronal populations [2]. |
| DAPI (4',6-diamidino-2-phenylindole) | Membrane-impermeant DNA dye for identifying intact nuclei during flow cytometry and assessing viability [2]. | Added to nuclei suspension prior to sorting to gate on DNA-containing particles [2]. |
| Low-Bind Tubes and Tips | Minimizes loss of nuclei and nuclear RNA due to surface adhesion, critical for low-input samples [33]. | Used for all steps involving nuclei suspension [33]. |
Experimental Workflow and Decision Pathways
The following workflow diagram synthesizes the key procedural steps and decision points for implementing an effective debris and myelin reduction strategy.
Figure 1: Optimized experimental workflow for reducing debris and myelin contamination during nuclei isolation from frozen brain tissue. The key decision point involves assessing myelin contamination levels to determine whether density gradient centrifugation is necessary.
Effective reduction of debris and myelin contamination is not merely a technical consideration but a fundamental prerequisite for generating high-quality data from single-nucleus genomic and epigenomic studies of frozen postmortem brain tissue. The protocols and comparative analyses presented herein provide a structured framework for researchers to optimize their nuclei isolation workflows. By implementing these targeted strategies—including optimized homogenization, strategic filtration, density-based myelin removal, and optional fluorescence-activated sorting—scientists can significantly improve nuclei integrity and purity, thereby ensuring the biological fidelity of their downstream analyses and advancing our understanding of neural mechanisms in health and disease.
Improving Nuclei Integrity and RNA Quality
Epigenetic and transcriptomic analyses of brain tissue provide crucial insights into neurodevelopment, aging, and neurodegenerative diseases [2]. For cellularly heterogeneous tissues like the brain, these investigations often require cell type-specific resolution, achieved through methods such as single-nucleus RNA sequencing (snRNA-seq) and assay for transposase-accessible chromatin with sequencing (ATAC-seq) [2] [3]. However, many protocols perform optimally with freshly harvested tissue, whereas inaccessible tissues like the brain are often obtained as frozen postmortem specimens, particularly from humans and non-human primates [2]. These samples present unique challenges, including high levels of myelin debris, reduced RNA integrity, and potential loss of disease-specific molecular signatures due to postmortem interval (PMI) [2] [35]. This application note details optimized protocols and critical considerations for isolating high-quality nuclei and preserving RNA integrity from frozen postmortem brain tissue, enabling reliable downstream single-cell and bulk genomic analyses.
Technical Challenges in Postmortem Brain Tissue Processing
Isolating nuclei from frozen postmortem brain tissue involves navigating several technical hurdles that can compromise yield, integrity, and downstream data quality.
- Inherent Tissue Properties: Primate brains possess a higher proportion of white matter than rodent brains, leading to elevated myelin debris during homogenization [2]. Furthermore, the postmortem collection of tissue from large animals and humans often experiences longer delays, exacerbating RNA degradation [2].
- Postmortem Interval (PMI) Effects: The time between death and tissue preservation (PMI) significantly impacts molecular integrity. While basic quality metrics like nuclei count and RNA Integrity Number (RIN) may remain stable, a prolonged PMI can obscure disease-specific transcriptomic signatures. A study comparing 0-hour and 3-hour PMI in mouse brains found that the 3-hour interval diminished the number of differentially expressed genes between disease (PS19) and wild-type models, particularly affecting stress and immune response pathways in neurons [35].
- Disease-Associated RNA Degradation: Evidence suggests that neurodegenerative diseases themselves can affect RNA quality. Research indicates a statistically significant decrease in RIN in Alzheimer's disease tissue compared to neurologically normal cases, a factor that must be accounted for in gene expression studies [36].
- Protocol-Dependent Biases: The choice of nuclei isolation method significantly influences experimental outcomes. Different protocols can alter cell type proportions, transcriptional homogeneity, and levels of ambient RNA contamination in snRNA-seq data [3]. For instance, column-based methods may result in substantial aggregation and debris, whereas sucrose gradient and machine-assisted methods typically yield purer nuclei suspensions [3].
Optimized Experimental Protocols
Protocol 1: Isolation from Frozen Primate Cerebral Cortex
This protocol, optimized for ~25 mg of frozen chimpanzee cerebral cortex tissue, is ideal for small, precious samples and includes steps for neuronal nuclei enrichment [2].
Detailed Methodology:
- Tissue Dissection: Hemisectioned whole brain slabs stored at -80°C should be microdissected on dry ice using a 2 mm biopsy punch. Weigh ~25-50 mg of frozen tissue per reaction in pre-chilled 1.5 mL microcentrifuge tubes [2].
- Nuclei Isolation: Use the 10X Genomics Chromium Nuclei Isolation Kit with RNase Inhibitor. The key optimizations include:
- Modified Lysis: Adjust the lysis time empirically to balance complete tissue disruption and nuclear membrane integrity.
- Enhanced Filtration: Pass the homogenate through a Flowmi cell strainer (40 µm) to remove large debris.
- Additional Washes: Incorporate extra wash steps with nuclei washing buffer to reduce myelin and RNA contamination [2].
- Quantification: Quantify nuclei suspension using an automated cell counter (e.g., Countess 3 FL). Mix 5 µL of nuclei suspension with 5 µL of viability stain (e.g., Acridine Orange/Propidium Iodide). A successful isolation will have a majority of Propidium Iodide-positive nuclei [2].
- Immunostaining & FANS (Optional):
- Subdivide the nuclei suspension for controls (unstained, secondary antibody only) and the sample.
- Incubate the sample with a primary antibody against the neuronal marker NeuN for 30 minutes on ice.
- Pellet nuclei by centrifugation (5 min at 400 rcf), remove supernatant, and resuspend in blocking buffer.
- Add a fluorescently-labeled secondary antibody and incubate for 15 minutes in the dark on ice.
- Wash twice to remove unbound antibody.
- Perform Fluorescence-Activated Nuclei Sorting (FANS) using a flow cytometer (e.g., Sony SH800Z). Use DAPI to gate all nuclei and sort the NeuN-positive population for neuronal enrichment [2].
Downstream Applications: Sorted neuronal nuclei are suitable for bulk epigenomic methods (e.g., methylome sequencing with enzymatic conversion) or single-cell library prep (ATAC-seq, RNA-seq). For low DNA inputs, use kits like the QIAamp DNA Micro Kit [2].
Protocol 2: A Simplified, Versatile Preparation for Multiple Tissues
This fast, low-cost protocol is optimized for ~20-50 mg of long-term frozen brain tumor tissue but has been successfully applied to various cryopreserved tissues [12] [5].
Detailed Methodology:
- Homogenization: Mince the frozen tissue in ice-cold lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl₂, 0.05% NP-40) on a pre-cooled surface. Transfer the tissue to a Dounce homogenizer.
- Critical Optimization: The number of strokes and the choice of pestle (loose: A, tight: B) must be tailored to the tissue type to optimize yield versus debris [5].
- Lysis and Filtration: Incubate the homogenate on ice for 5 minutes. Stop the lysis by adding a large volume of ice-cold nuclei washing buffer (PBS, BSA, Glycerol, RNase inhibitor). Filter the suspension through a 30 µm cell strainer [12] [5].
- Debris Removal and Concentration: Centrifuge the filtrate at 500 rcf for 10 minutes at 4°C. Resuspend the pellet in nuclei washing buffer. For further purification, layer the suspension over a 29% iodixanol cushion and centrifuge at 1000 g for 10 minutes. The intact nuclei will form a pellet, separated from the debris [12].
- Nuclei Sorting (Recommended for Purity): Stain the nuclei suspension with 7-AAD viability dye. Sort nuclei using a flow sorter (e.g., BD FACSAria Fusion) to collect intact, fluorescent-positive events. This step dramatically reduces ambient RNA and cellular debris, ensuring a high-quality input for snRNA-seq [5].
Workflow Diagram
The following diagram illustrates the key decision points and steps in the two optimized protocols.
Comparative Performance of Isolation Methods
The choice of nuclei isolation method is a critical experimental variable that directly impacts nuclei yield, integrity, and the biological interpretation of snRNA-seq data [3]. A systematic comparison of three mechanistically distinct strategies highlights their relative performance.
Table 1: Quantitative Comparison of Nuclei Isolation Methods from Mouse Brain Cortex
| Method | Approximate Yield per mg Tissue | Nuclei Integrity | Relative Debris Level | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Sucrose Gradient Centrifugation [3] | ~60,000 | 85% | Low | Well-established, cost-effective | Person-to-person variability, requires ultracentrifuge |
| Spin Column-Based [3] | ~45,000 | 35% | High | Fast processing, no special machinery | Low integrity, high aggregation and debris |
| Machine-Assisted Platform [3] | ~60,000 | ~100% | Negligible | High throughput, minimal variability, excellent integrity | Requires specialized equipment and consumables |
The data show that while the sucrose gradient and machine-assisted methods provide superior yield and integrity, the column-based method underperforms, yielding only 35% intact nuclei with substantial debris [3]. Furthermore, the isolation technique influences the cell types captured; for example, the sucrose gradient method captured the largest proportion of astrocytes (13.9%), while the machine-assisted method best preserved oligodendrocyte (15.9%) and microglia (5.6%) populations [3].
The Scientist's Toolkit: Essential Reagents and Materials
Successful isolation of high-integrity nuclei relies on specific reagents and equipment to maintain RNA quality and nuclear integrity.
Table 2: Key Research Reagent Solutions for Nuclei Isolation
| Item | Function/Application | Example Products & Kits |
|---|---|---|
| Nuclei Isolation Kit | Provides optimized buffers for lysis and stabilization. | Chromium Nuclei Isolation Kit (10X Genomics) [2], Nuclei EZ Prep (Sigma-Aldrich) [12] |
| RNase Inhibitor | Prevents degradation of nuclear RNA during isolation. | Protector RNase Inhibitor [5] |
| Density Gradient Medium | Purifies nuclei by separating them from cellular debris based on density. | Iodixanol (OptiPrep) [12] [5], Sucrose solutions [3] |
| Antibodies for FANS | Enables isolation of specific nuclear populations (e.g., neuronal). | Anti-NeuN (Neuronal nuclei) [2], Anti-Erg (Endothelial nuclei) [37] |
| Viability/ Nucleic Acid Stains | Distinguishes intact nuclei and facilitates sorting or quantification. | DAPI [2], Propidium Iodide [2], 7-AAD [5] |
| Homogenization Equipment | Mechanically disrupts tissue to release nuclei while minimizing damage. | Dounce Homogenizer (loose & tight pestles) [5], Biopsy Punch (for microdissection) [2] |
Isolating high-quality nuclei from frozen postmortem brain tissue is a foundational step for reliable single-cell and bulk genomic analyses. The protocols and data presented herein demonstrate that success hinges on a multifaceted approach: selecting an isolation method matched to experimental goals (e.g., sucrose gradient for cost-effectiveness, FANS for purity), meticulously controlling for pre-analytical variables like PMI and disease state, and incorporating key optimizations such as tailored homogenization and debris-removal steps. By adhering to these refined protocols, researchers can maximize the scientific return from precious and irreplaceable postmortem brain samples, paving the way for deeper insights into the cellular mechanisms of brain function and disease.
Maximizing Yield from Small or Precious Samples
The molecular analysis of frozen postmortem brain tissue is fundamental for advancing research in neurobiology, disease mechanisms, and drug development. However, such samples, particularly from non-human primates (NHPs) and humans, are often irreplaceable and available in limited quantities, making the maximization of nuclei yield and quality a critical laboratory procedure [2] [38]. The challenges are multifaceted: postmortem intervals can reduce RNA integrity, primate brains contain high levels of myelin debris that complicate purification, and standard protocols often require impractically large starting amounts of tissue [2]. Furthermore, the choice of nuclei isolation method has been demonstrated to significantly impact cell type proportions, transcriptional data quality, and the overall biological interpretation of single-nucleus RNA-sequencing (snRNA-seq) experiments [3]. This application note provides a consolidated guide of optimized protocols and analytical comparisons, framed within a broader thesis on nuclei isolation, to empower researchers to extract the maximum scientific value from small or precious frozen brain samples.
Comparative Analysis of Nuclei Isolation Methods
Selecting an appropriate nuclei isolation strategy is the first and most crucial step in ensuring a successful downstream analysis. Different methods offer distinct trade-offs between yield, purity, required equipment, and suitability for various sample types. The following table summarizes the key characteristics of several prominent methods, enabling an informed choice based on specific research constraints and goals.
Table 1: Comparison of Nuclei Isolation Methods for Brain Tissue
| Method | Key Features & Modifications | Recommended Tissue Input | Reported Performance | Best Suited For |
|---|---|---|---|---|
| Optimized Sucrose Gradient Centrifugation [3] | Manual homogenization (Dounce) followed by sucrose gradient ultracentrifugation. | ~30 mg [3] | High yield (~60,000 nuclei/mg) 85% nuclei integrity Captures diverse cell types, including a high proportion of astrocytes (13.9%) [3] | Projects requiring high nuclei integrity and comprehensive cell type representation, where ultracentrifugation is accessible. |
| Commercial Kit with Optimized Lysis [2] [12] | Use of 10X Genomics Nuclei Isolation Kit, optimized with adjusted lysis time, extra washes, and added filtration steps. | ~25 mg [2] | Good yield from small inputs Reduced debris and background fluorescence Compatible with FANS and single-cell protocols [2] | Low-input NHP/human postmortem samples; labs with standard equipment seeking a robust, optimized workflow. |
| Iodixanol Density Gradient [5] | Homogenization followed by purification on a 29% iodixanol cushion and nuclei sorting. | As low as 15 mg [5] | High purity Effective debris removal Successfully profiled 1,500–7,500 nuclei per tissue from multiple cancer types [5] | Very low-input clinical samples and biobanked cryopreserved tissues where maximum sample purity is essential. |
| Simplified & Fast Protocol [12] | A fast (<30 min), low-cost method relying on douncing, filtration, and wash steps without a density gradient. | 20–50 mg [12] | Good balance of yield and purity Low mitochondrial reads (<1%) in sequencing data Simplicity and cost-effectiveness [12] | Long-term frozen brain tumor tissues; labs prioritizing a simple, rapid, and economical protocol. |
Detailed Experimental Protocols
Optimized Protocol for Frozen Postmortem Primate Cortex
This protocol, adapted from Guevara et al. (2025), is specifically tailored for small amounts of frozen chimpanzee cerebral cortex and includes steps for neuronal nuclei enrichment [2] [38].
Workflow Overview
Diagram 1: Workflow for primate cortex nuclei isolation and sorting.
Step-by-Step Methodology
- Tissue Dissection: On dry ice, use a biopsy punch to microdissect ~25 mg of frozen cerebral cortex tissue. Perform all subsequent steps on ice or with pre-chilled equipment [2].
- Nuclei Isolation: Use a commercial nuclei isolation kit (e.g., 10X Genomics Chromium Nuclei Isolation Kit) as a base. Critically, optimize the lysis time and incorporate additional wash steps to reduce myelin debris. Pass the nuclei suspension through a pre-chilled 40 µm Flowmi cell strainer [2].
- Quantification and Viability: Mix 5 µL of nuclei suspension with 5 µL of a viability stain (e.g., Acridine Orange/Propidium Iodide). Quantify using an automated cell counter. A successful isolation will show a majority of Propidium Iodide-positive (RFP-labeled) nuclei [2] [39].
- Immunostaining for FANS (Optional):
- Subdivide the nuclei suspension into controls (unstained, secondary antibody only) and the sample.
- Incubate the sample with a primary antibody against the neuronal marker NeuN for 30 minutes on ice.
- Pellet nuclei by centrifugation (400 rcf, 5 min), remove supernatant, and resuspend in blocking buffer.
- Add a fluorescently-labeled secondary antibody and incubate for 15 minutes in the dark on ice. Wash twice to remove unbound antibody [2].
- Fluorescent-Activated Nuclei Sorting (FANS):
- Keep samples on ice. Add DAPI to the suspension shortly before sorting.
- Use an unstained control to set voltages and a DAPI gate to identify intact nuclei.
- Use the secondary antibody-only control to set the background threshold for the fluorescence channel.
- Sort the DAPI-positive and NeuN-positive (for neuronal enrichment) populations. Collect sorted nuclei in tubes containing chilled blocking buffer [2].
- Downstream Applications: Sorted nuclei can be used for bulk methylome sequencing (using kits optimized for low DNA input, e.g., QIAamp DNA Micro Kit) or for single-cell library preparation (e.g., 10X Genomics) [2].
Simplified Protocol for Long-Term Frozen Brain Tumor Tissue
This protocol, from Scientific Reports (2025), is designed for simplicity and cost-effectiveness, requiring less than 30 minutes to complete [12].
Workflow Overview
Diagram 2: Simplified workflow for frozen brain tumor tissue.
Step-by-Step Methodology
- Homogenization: Mince approximately 30 mg of frozen tissue in ice-cold lysis buffer (e.g., 10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl₂, 0.05% NP-40) using a scalpel. Transfer the mixture to a Dounce homogenizer. Perform homogenization with a loose pestle (Pestle A) for about 10-15 strokes, followed by a tight pestle (Pestle B) for 10-15 strokes, on ice [5] [12].
- Filtration and Washing: Filter the homogenate through a 30-40 µm MACS strainer to remove large debris. Centrifuge the filtrate at 1000 g for 10 minutes at 4°C. Carefully remove the supernatant.
- Critical Wash Steps: Resuspend the pellet in lysis buffer without detergent and incubate on ice for 5 minutes. This step helps remove cytoplasmic debris without permeabilizing the nuclear walls. Centrifuge again and repeat the wash 1-2 more times. Note: While three washes are optimal for purity, each wash causes nuclei loss. For very low-input samples, two washes may be preferable [12].
- Final Resuspension and Storage: Resuspend the final pellet in an appropriate nuclei storage or washing buffer (e.g., containing PBS, BSA, glycerol, and RNase inhibitor). The nuclei can be used immediately for snRNA-seq or frozen at -80°C for a short period (2-3 days) [5] [12].
The Scientist's Toolkit: Essential Research Reagents and Materials
The success of nuclei isolation from challenging samples often depends on the specific reagents and tools used. The following table catalogues key solutions mentioned in the optimized protocols.
Table 2: Key Research Reagent Solutions for Nuclei Isolation
| Item | Function/Application | Specific Example/Note |
|---|---|---|
| Dounce Homogenizer | Mechanical tissue disruption to release nuclei while minimizing damage. | Pestle clearance is critical: "loose" (0.0025-0.0055 in) and "tight" (0.0005-0.0025 in) pestles are used sequentially [5] [12]. |
| Nuclei Isolation Buffer | Provides an isotonic environment for lysis and stabilization. | Often contains Tris-HCl, NaCl, MgCl₂, and a non-ionic detergent (e.g., NP-40). Detergent-free versions are used for washing [5] [12]. |
| Density Gradient Media | Purifies nuclei by separating them from dense cellular debris. | Sucrose cushions or Iodixanol (Optiprep) gradients are common. Choice affects yield and purity [3] [5]. |
| Fluorescent Viability Stains | Differentiating intact nuclei from debris and assessing quality. | Propidium Iodide (PI) and 7-AAD are membrane-impermeable and stain DNA in nuclei. Acridine Orange (AO) stains all nucleic acids [2] [5] [39]. |
| Antibodies for FANS | Enrichment of specific cell populations (e.g., neurons). | Anti-NeuN (Neuronal Nuclei) antibody is widely used for neuronal enrichment from brain nuclei suspensions [2]. |
| RNase Inhibitors | Preserve nuclear RNA integrity during the isolation process. | Essential for all steps post-homogenization to prevent RNA degradation, especially in postmortem samples [2] [5]. |
| MACS Strainers (30-40µm) | Remove large aggregates and tissue clumps post-homogenization. | A critical step for reducing background debris in the final suspension [5] [12]. |
Maximizing the yield and quality of nuclei from small or precious frozen brain samples is an achievable goal through the implementation of carefully optimized and validated protocols. The key lies in understanding the specific challenges of the sample type—whether it's NHP cortex with high myelin content or long-term frozen tumor tissue—and selecting a method that balances yield, purity, and practical constraints. By integrating strategic modifications such as optimized lysis conditions, additional filtration and wash steps, and optional fluorescence-activated sorting, researchers can significantly enhance the quality of their downstream genomic data. This ensures that every milligram of these invaluable samples is utilized to its fullest potential, driving robust and reproducible scientific discovery.
FACS Gate Setting and Purity Validation
Within the context of a broader thesis on nuclei isolation from frozen postmortem brain tissue, establishing a rigorous Fluorescence-Activated Cell Sorting (FACS) gating strategy and purity validation protocol is paramount. The integrity of downstream genomic and epigenomic analyses, such as single-nuclei RNA sequencing (snRNA-seq) and methylome profiling, is entirely contingent upon the initial purity and accurate identification of isolated nuclei populations [2]. This application note provides a detailed protocol for setting FACS gates and validating sorted nuclei purity, specifically optimized for the unique challenges presented by frozen postmortem brain tissue, which is characterized by high levels of intrinsic debris, reduced RNA integrity, and autofluorescence [2].
Critical Pre-Sorting Considerations for Nuclei
Nuclei Isolation from Frozen Postmortem Brain Tissue
The initial nuclei isolation step is critical for the success of all subsequent FACS and analytical procedures. For frozen postmortem chimpanzee cerebral cortex tissue, an optimized protocol has been developed for small starting amounts (~25 mg) [2]. The process involves microdissection on dry ice, followed by tissue homogenization using a Dounce homogenizer with pestles of specific clearances (loose: 0.0025–0.0055 inches; tight: 0.0005–0.0025 inches) [2]. The lysis buffer typically consists of 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl₂, and 0.05% NP-40, with lysis time being a key optimized variable [2]. Following homogenization, enhanced filtration through 30 µm MACS strainers and additional wash steps are incorporated to reduce debris [2]. For purification, a density gradient centrifugation step using iodixanol (Optiprep) can be employed to isolate intact nuclei from myelin and other cellular debris [2] [5].
Key Research Reagent Solutions
The following reagents are essential for successful nuclei isolation, staining, and sorting.
Table 1: Essential Reagents for Nuclear Isolation and FANS
| Reagent Category | Specific Examples | Function |
|---|---|---|
| Nuclei Isolation | Dounce Homogenizer & Pestles, NP-40 Lysis Buffer, 30 µm MACS Strainers, Iodixanol (Optiprep) | Mechanical tissue disruption, cell membrane lysis, removal of large debris, and purification of nuclei via density gradient. |
| Viability/ Nucleic Acid Staining | DAPI, Propidium Iodide (PI), 7-AAD, Acridine Orange (AO) | Membrane-impermeant dyes (DAPI, PI, 7-AAD) stain DNA in intact nuclei, allowing for viability gating. AO stains nucleic acids in both cells and nuclei [2] [40]. |
| Immunostaining | Primary Antibody (e.g., Anti-NeuN), Fluorophore-conjugated Secondary Antibody, Blocking Buffer | Immunological identification of specific nuclear populations (e.g., neuronal nuclei via NeuN) for targeted sorting [2]. |
| Controls | Unstained Nuclei, Secondary Antibody Only Control, Fluorescence Minus One (FMO) Controls | Determining background fluorescence, assessing antibody specificity, and establishing accurate positive gates [41] [2]. |
A Sequential FACS Gating Strategy for Nuclei
A step-by-step gating strategy is essential to isolate a pure population of intact nuclei.
Figure 1: Sequential FACS Gating Strategy for Nuclei Purity. This workflow outlines the logical progression of gates to isolate a specific, pure population of nuclei.
Gate 1: Exclusion of Debris and Selection of Intact Nuclei
The first plot created is Forward Scatter-Area (FSC-A) versus Side Scatter-Area (SSC-A). FSC correlates with particle size, while SSC indicates internal complexity or granularity [41] [40]. On this plot, intact nuclei will form a distinct population that is separate from smaller debris particles (which are FSC-low and SSC-low) and larger, more complex aggregates [41]. A region (e.g., "R1 - Intact Nuclei") is drawn around the population of interest.
Gate 2: Exclusion of Doublets and Multiplets
To ensure that each data point corresponds to a single nucleus, it is crucial to exclude doublets (two nuclei stuck together) or multiplets. This is achieved by creating a plot of FSC-Height (FSC-H) versus FSC-A and applying the previous "Intact Nuclei" gate to it [41]. As they pass through the laser, single nuclei will have a proportional relationship between their height and area signals, forming a diagonal line. Doublets will have a disproportionately large FSC-A value compared to their FSC-H, appearing above the main population [41]. A region is drawn around the singlet population (e.g., "R2 - Single Nuclei").
Gate 3: Selection of Viable Nuclei
Nuclei integrity is assessed using membrane-impermeant viability dyes that intercalate with DNA, such as DAPI, Propidium Iodide (PI), or 7-AAD [2] [40]. A plot of SSC-A versus the viability dye channel is created, and the "Single Nuclei" gate is applied. Intact nuclei, which have permeable nuclear membranes but intact surrounding membranes, will be positive for the viability dye [2]. A region is drawn around this positive population (e.g., "R3 - Viable Nuclei").
Gate 4: Selection of Target Population
The final gating step identifies the specific nuclear population of interest based on immunostaining or other fluorescent markers. For example, to isolate neuronal nuclei, a primary antibody against the neuronal marker NeuN is used, followed by a fluorophore-conjugated secondary antibody [2]. A plot of the relevant fluorophore channel is used, with all previous gates applied. The positive population is defined using appropriate controls.
Establishing Accurate Positive Gates with Appropriate Controls
Proper use of controls is non-negotiable for distinguishing true positive signals from background and spillover.
Table 2: Key Controls for FACS Gate Setting
| Control Type | Composition | Primary Function | Gating Application |
|---|---|---|---|
| Unstained Control | Nuclei suspension without any antibodies or dyes. | To assess autofluorescence and set baseline detector voltages. | Determines the negative population for all channels [2]. |
| Fluorescence Minus One (FMO) Control | Contains all fluorophore-labeled antibodies except one. | To account for background fluorescence and spectral spillover into the channel of the omitted antibody in a multicolor panel. | Critical for accurately setting the boundary between negative and positive populations, especially for low-abundance markers [41]. |
| Isotype Control | An antibody with the same isotype but irrelevant specificity, matched for fluorophore and concentration. | To assess nonspecific antibody binding. | Can help set gates for specificity, though its limitations are recognized [41]. |
| Biological Control | A cell subset in the test sample known not to express the target marker. | Provides a biological negative control. | The most robust method for defining a true negative population [41] [42]. |
It is critical to note that isotype controls only address non-specific antibody binding and not fluorescence spillover, and FMO controls only address spillover [42]. Therefore, the use of well-defined biological controls (e.g., a non-target nuclear population) is considered the gold standard for proper gate placement [42].
Post-Sort Purity Validation and Downstream Applications
Direct Analysis of Sorted Populations
The most direct method for validating sort purity is to re-analyze a small aliquot of the sorted "positive" population on the flow cytometer. The percentage of events that fall within the original positive gate should typically exceed 95% for high-purity sorts. This confirms that the gating strategy was effective during the sort.
Functional Validation via Downstream Assays
The ultimate validation of nuclei purity and integrity is their performance in functional genomic assays.
- Single-Cell Genomics: Sorted neuronal (NeuN+) and non-neuronal (NeuN-) nuclei populations can be used to generate 10X Genomics 3'-RNA-seq libraries. Successful sequencing should show distinct gene expression profiles, with neuronal nuclei expressing markers such as SNAP25 and SYT1, while non-neuronal nuclei express markers like GFAP (astrocytes) or MOBP (oligodendrocytes) [2]. This functionally validates the sort purity.
- Bulk Epigenomic Analysis: For bulk methylome sequencing, DNA is extracted from sorted neuronal nuclei using a kit optimized for low inputs (e.g., QIAamp DNA Micro Kit) [2]. Enzymatic methyl-seq (e.g., using the NEBNext Enzymatic Methyl-seq Kit) is recommended over bisulfite conversion for low-DNA samples as it causes less DNA damage [2]. Successful sequencing with low PCR duplication rates and sufficient genome-wide CpG coverage confirms the quality of the sorted nuclei.
A methodical approach to FACS gate setting and validation is the foundation for obtaining high-quality data from isolated nuclei. This involves a sequential gating strategy to isolate intact, single, and viable nuclei, complemented by the stringent use of FMO and biological controls to define positive populations accurately. Finally, post-sort purity analysis and functional validation in downstream assays confirm that the sorted nuclei populations are of sufficient purity and quality for robust single-cell or bulk genomic and epigenomic analysis. This comprehensive protocol ensures that precious samples, such as frozen postmortem brain tissue, are utilized to their fullest potential.
Validation and Quality Control: Ensuring Data Integrity and Biological Relevance
Microscopy and Flow Cytometry for Quality Assessment
Within the broader context of establishing a robust protocol for nuclei isolation from frozen postmortem brain tissue, quality assessment is a critical, non-negotiable step. The success of downstream genomic and transcriptomic applications—such as single-nucleus RNA sequencing (snRNA-seq) and methylome analysis—depends entirely on the initial integrity and purity of the isolated nuclei [2] [3]. For cellularly heterogeneous tissues like the brain, where intricate networks of neurons and glia perform specialized functions, cell type-specific analysis is often required to uncover meaningful biological insights [2] [43]. This Application Note details the essential microscopy and flow cytometry techniques used to evaluate nuclei quality, providing a structured framework to ensure that isolated nuclei populations meet the stringent standards necessary for reliable and reproducible research outcomes.
Quantitative Assessment of Nuclei Quality
Initial quality control provides a quantitative foundation for deciding whether a nuclei preparation is suitable for downstream applications. Key metrics include nuclei yield, integrity, and the level of contaminating debris.
Table 1: Key Quantitative Metrics for Nuclei Quality Assessment
| Metric | Description | Measurement Technique | Significance for Downstream Applications |
|---|---|---|---|
| Nuclei Yield | Total number of nuclei recovered per milligram of input tissue. | Automated cell counters (e.g., Countess 3 FL) or flow cytometry with counting beads [2] [44]. | Determines the scale of possible experiments; low yield may preclude certain protocols. |
| Nuclei Integrity/Viability | Percentage of nuclei with intact membranes. | Fluorescent viability stains (e.g., Acridine Orange/Propidium Iodide or DAPI); intact nuclei stain positively [2] [3]. | Ensures RNA/DNA integrity and reduces background signal from lysed nuclei. |
| Debris Level | Amount of non-nuclear particulate matter in the suspension. | Brightfield microscopy and flow cytometry side-scatter (SSC) analysis [3]. | High debris can clog microfluidic devices (e.g., 10X Chromium) and impede accurate sorting. |
The choice of isolation protocol profoundly impacts these quantitative outcomes. A recent comparative analysis of three mechanistically distinct nuclei isolation methods for brain tissue revealed significant differences in performance (Table 2). This underscores the importance of method selection and rigorous quality control.
Table 2: Comparative Performance of Nuclei Isolation Methods for Mouse Brain Cortex [3]
| Isolation Method | Approximate Yield (per mg tissue) | Nuclei Integrity | Observed Debris | Notable Characteristics |
|---|---|---|---|---|
| Sucrose Gradient Centrifugation | ~60,000 | 85% | Minimal | Well-established, cost-effective; potential for person-to-person variability. |
| Spin Column-Based | ~25% fewer than other methods | 35% | Substantial aggregation and debris | Faster processing; lower yield and integrity may limit utility. |
| Machine-Assisted Platform | ~60,000 | ~100% | Negligible | Automated, highly reproducible; requires specialized, costly equipment. |
Detailed Experimental Protocols
Protocol 1: Quantification and Viability Staining Using Fluorescent Microscopy
This protocol is adapted for use with an automated cell counter like the Countess 3 FL, providing a rapid assessment of nuclei concentration and integrity [2].
Materials:
- Nuclei suspension (from isolation protocol)
- ReadyCount Red/Green Viability Stain (Thermo Fisher, #A49905) or equivalent containing Acridine Orange (AO) and Propidium Iodide (PI)
- Microcentrifuge tubes (0.2 mL and 1.5 mL)
- Pipettes and tips
Procedure:
- Prepare Stain Mixture: Gently mix the nuclei suspension by pipetting. Combine 5 µL of the nuclei suspension with 5 µL of ReadyCount Red/Green Viability Stain in a 0.2 mL tube. Pipette up and down to mix thoroughly.
- Load and Analyze: Pipette the entire 10 µL mixture into a counting chamber slide. Insert the slide into the automated cell counter.
- Interpret Results: The counter will identify and count nuclei based on fluorescence.
- AO (green) is membrane-permeant and stains all nuclei.
- PI (red) is membrane-impermeant and only stains nuclei with compromised membranes.
- A successful isolation will show a high percentage of nuclei that are double-stained or labeled only with PI, indicating a population of intact, membrane-bound nuclei [2].
Protocol 2: Cell Population Estimation via Flow Cytometry (Flow Fractionator)
This method, known as the "flow fractionator," enables high-throughput, precise estimation of total cell numbers in homogenized brain tissue and is well-suited for processing hundreds of samples [44].
Materials:
- Nuclei suspension
- DAPI (4′,6-diamidino-2-phenylindole) stock solution
- CountBright Absolute Counting Beads (Invitrogen)
- Flow Cytometer equipped with a 355 nm laser (e.g., BD LSR II)
- PBS (Phosphate Buffered Saline)
Procedure:
- Sample Preparation: Thoroughly vortex the main nuclei suspension. Combine a 50 µL aliquot of the nuclei suspension with 250 µL of a DAPI-PBS solution and 50 µL of CountBright counting beads.
- Flow Cytometer Setup: Use a flow cytometer with a 355 nm laser for DAPI excitation. Create a two-parameter plot of Side Scatter (SSC-A) versus DAPI fluorescence (DAPI-A).
- Gating and Acquisition:
- Place a selection gate around the population of DAPI-positive events, excluding small debris based on the SSC-A and DAPI-A signals [44].
- Acquire data until 1,000 bead events are recorded. The absolute number of nuclei in the sample is calculated based on the known concentration of the counting beads and the ratio of nuclei events to bead events.
- Quality Control: Prepare and run samples in duplicate to assess technical variation. The coefficient of variation between replicates should be low for reliable data.
Protocol 3: Immunostaining and Fluorescence-Activated Nuclei Sorting (FANS)
This protocol allows for the specific enrichment of neuronal nuclei (NeuN+) from a heterogeneous nuclei suspension, enabling cell type-specific downstream analysis [2].
Materials:
- Primary Antibody: Anti-NeuN (Neuronal Nuclei)
- Fluorescently-labeled Secondary Antibody
- Blocking Buffer (e.g., 1% BSA in PBS)
- DAPI Stain (#D1306, Thermo Fisher)
- Pre-chilled microcentrifuge tubes
- Fluorescent-Activated Cell Sorter (e.g., Sony SH800Z)
Procedure:
- Aliquot and Stain: Subdivide the quantified nuclei suspension into separate tubes for: a) unstained control, b) secondary antibody-only control, and c) the experimental sample. Pellet the nuclei for the experimental sample by centrifugation at 400 rcf for 5 min, then resuspend in blocking buffer containing the primary Anti-NeuN antibody. Incubate for 30 minutes on ice.
- Wash and Secondary Stain: Pellet nuclei again to remove unbound primary antibody. Resuspend the pellet in blocking buffer and add the appropriate fluorescently-labeled secondary antibody. Incubate for 15 minutes in the dark on ice.
- Final Wash: Pellet nuclei and wash twice with blocking buffer to remove unbound secondary antibody. Keep samples on ice and protected from light.
- Flow Cytometry Controls and Gating:
- Setup: Run the unstained control to set photomultiplier tube (PMT) voltages and establish a negative fluorescence gate.
- DAPI Gate: Add DAPI to the unstained control and run to establish a gate for all nucleated events (DAPI-positive).
- Background Control: Add DAPI to the secondary antibody-only control to set the background threshold for the fluorescence channel of the secondary antibody.
- Sample Sorting: Add DAPI to the experimental sample. Sort the nuclei that are within the DAPI gate and exhibit fluorescence above the background threshold in the secondary antibody channel (NeuN+). Collect sorted neuronal nuclei in tubes containing chilled blocking buffer for immediate use or nucleic acid extraction [2].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Quality Assessment
| Item | Function | Example Product/Catalog Number |
|---|---|---|
| DAPI (4′,6-diamidino-2-phenylindole) | DNA intercalating dye that stains all nuclei; used for total nuclei counting and viability assessment. | Thermo Fisher D1306 [2] [44] |
| Acridine Orange/Propidium Iodide Stain | Dual-fluorescence viability stain for distinguishing intact (AO+/PI-) from compromised nuclei. | Invitrogen ReadyCount Red/Green (#A49905) [2] |
| Absolute Counting Beads | Precisely quantified beads for determining absolute nuclei counts in flow cytometry. | Invitrogen CountBright Absolute Counting Beads [44] |
| Anti-NeuN Antibody | Primary antibody that binds to a neuronal-specific nuclear protein, enabling neuronal enrichment. | Millipore Sigma MAB377 [2] [45] |
| Chromium Nuclei Isolation Kit | Commercial kit for isolating nuclei from small amounts of frozen tissue; a common starting point for optimized protocols. | 10X Genomics Nuclei Isolation Kit (PN-1000494) [2] |
| QIAamp DNA Micro Kit | DNA extraction kit optimized for low inputs, suitable for use with sorted nuclei populations. | Qiagen QIAamp DNA Micro Kit [2] |
Workflow Visualization
The following diagram illustrates the logical progression from nuclei isolation to final quality assessment and sorting, integrating the protocols described above.
Benchmarking Against Public Single-Cell and Single-Nuclei Atlases
Integrating data from public single-cell and single-nuclei atlases is becoming a critical step in the validation pipeline for studies utilizing nuclei isolated from frozen postmortem brain tissue. By benchmarking newly generated datasets against well-curated public references, researchers can authenticate cell type identities, assess data quality, and place their findings within the broader context of established molecular landscapes. This application note provides detailed methodologies for nuclei isolation from challenging frozen postmortem brain samples and a structured framework for benchmarking against public atlases, with a focus on the human hippocampus.
Experimental Protocols
Optimized Nuclei Isolation from Frozen Postmortem Brain Tissue
The quality of single-nucleus RNA sequencing (snRNA-seq) data is fundamentally dependent on the initial nuclei isolation. The following protocol is optimized for small amounts (25-50 mg) of frozen postmortem brain tissue, addressing challenges such as high myelin debris and reduced RNA integrity [2].
Detailed Protocol [2]:
- Tissue Dissection: On dry ice, microdissect frozen brain tissue slabs using a 2 mm biopsy punch. Weigh out 25-50 mg of tissue per isolation reaction.
- Initial Homogenization: Mince the weighed tissue in a pre-chilled NIM1 buffer. Centrifuge the suspension at 300 × g for 8 minutes. Wash the resulting pellet with PBS containing 1% BSA and centrifuge again under the same conditions.
- Dounce Homogenization: Resuspend the tissue pellet in a homogenization buffer (HB). Transfer to a Dounce homogenizer and perform 12-15 strokes with a tight-fitting pestle. Incubate the homogenate on ice for 10 minutes.
- Filtration and Debris Removal: Centrifuge the homogenate at low speed (300 rpm for 5 minutes) to pellet large debris. Filter the supernatant through a series of cell strainers with progressively smaller pore sizes (e.g., 100 μm, 70 μm, and 40 μm).
- Nuclei Collection: Centrifuge the filtered suspension at 300 × g for 10 minutes to pellet the nuclei. Resuspend the purified nuclei pellet in PBS with 1% BSA.
- Quality Control (QC): Assess nuclei integrity, concentration, and the extent of cytoplasmic contamination using an automated cell counter (e.g., Countess 3 FL) with a viability stain like Acridine Orange (AO)/Propidium Iodide (PI). A successful isolation will show a majority of intact, double-stained nuclei [2].
- Optional Fluorescent-Activated Nuclei Sorting (FANS): For neuronal enrichment, incubate the nuclei suspension with a primary antibody against the neuronal marker NeuN, followed by a fluorophore-conjugated secondary antibody. Use a flow cytometer to sort the NeuN-positive and NeuN-negative populations. Always include appropriate controls (unstained, secondary antibody only) for accurate gating [2].
Critical Steps and Troubleshooting [2]:
- Challenge: High myelin debris is common in primate brain tissue.
- Solution: Incorporate additional wash steps and optimized filtration. Sucrose density gradient ultracentrifugation can be used as an optional clean-up step.
- Challenge: Suboptimal lysis leading to low yield or poor nuclei integrity.
- Solution: Precisely optimize the lysis time during homogenization for the specific tissue type and postmortem interval.
- Challenge: Reduced RNA integrity number (RIN) in postmortem samples.
- Solution: Include RNase inhibitors in all buffers and work quickly on ice to preserve RNA quality.
Benchmarking Against Public Hippocampal Atlas Data
After generating snRNA-seq data from isolated nuclei, the following workflow allows for rigorous benchmarking against a public atlas, such as the integrated single-nucleus and spatial transcriptomics atlas of the human hippocampus [46].
Benchmarking Workflow [46]:
Data Acquisition and Preprocessing:
- Download the raw and processed data from the public atlas (e.g., NCBI GEO or the provided web application).
- Preprocess your newly generated snRNA-seq data (quality control, normalization, and scaling) to mirror the methods used in the reference atlas.
Cell Type Label Transfer and Integration:
- Utilize anchor-based integration methods (e.g., as implemented in Seurat or SCANPY) to co-embed your query dataset with the reference atlas.
- Perform label transfer, where previously annotated cell types from the reference are predicted for each cell in your new dataset. This validates the identity of your cell populations.
Spatial Inference with Non-negative Matrix Factorization (NMF):
- If the reference atlas includes spatially resolved transcriptomics (SRT) data, leverage the deconvolution and NMF factors generated from the integrated snRNA-seq data.
- Apply these factors to your data to infer the spatial organization of cell types and gene expression patterns, even from dissociated nuclei data.
Comparative and Differential Expression Analysis:
- Conduct pseudobulk-level comparisons between your data and the atlas. Use linear mixed-effects models to identify consistent and divergent differentially expressed genes (DEGs) across cell types and hippocampal subregions.
- Correlate gene-level enrichment statistics (e.g., t-statistics from differential expression models) between your dataset and the atlas to quantify reproducibility [46].
Functional and Circuit-Level Inference:
- Leverage the integrated human data and cross-species comparisons (e.g., with rodent functional data) from the atlas to generate hypotheses about activity-dependent transcription and circuit connectivity in your own dataset.
Table 1: Key Resources from a Public Human Hippocampus Atlas for Benchmarking [46]
| Resource Type | Description | Utility in Benchmarking |
|---|---|---|
| snRNA-seq Data | 75,411 high-quality nuclei from 10 neurotypical donors, annotated to 24 cell types. | Core reference for cell type label transfer and validation. |
| Spatial Transcriptomics | 150,917 Visium spatial spots from 36 capture areas across hippocampal subfields. | Enables inference of spatial localization for nuclei data. |
| Differential Expression | Marker genes for spatial domains (e.g., PPFIA2 for GCL, PRKCG for CA1-CA4) and neuronal subclusters. | Provides a gene set for validating cellular identity and data quality. |
| Web Applications | Interactive portals for exploring data. | Allows for rapid, visual cross-checking of gene expression patterns. |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Nuclei Isolation and snRNA-seq
| Item | Function | Example/Note |
|---|---|---|
| Nuclei Isolation Kit | Provides optimized buffers for lysis and stabilization of nuclei. | 10X Genomics Chromium Nuclei Isolation Kit; requires optimization for brain tissue [2]. |
| Cell Strainers | Removal of tissue debris and clumps after homogenization. | Use a series (100μm, 70μm, 40μm) for maximum debris removal [47]. |
| Dounce Homogenizer | Mechanical tissue disruption to liberate nuclei while preserving integrity. | Critical for consistent results; prefer glass over plastic. |
| RNase Inhibitor | Protects RNA from degradation during the isolation procedure. | Essential for postmortem tissue with potentially lower RNA integrity [2]. |
| Viability Stain | Distinguishes intact nuclei from debris and dead cells. | Acridine Orange (AO)/Propidium Iodide (PI); PI+ nuclei indicate successful lysis [2]. |
| Anti-NeuN Antibody | Immunostaining for fluorescence-activated nuclei sorting (FANS) to enrich neuronal nuclei. | Enables cell-type-specific analyses or bulk methylome sequencing from neurons [2]. |
| Single-Cell Library Kit | Preparation of barcoded cDNA libraries for sequencing. | 10X Genomics 3' Gene Expression; compatible with isolated nuclei. |
| Bovine Serum Albumin (BSA) | Reduces non-specific binding in buffers, improving sorting efficiency. | Use at 1% in PBS for wash and resuspension buffers [2]. |
Workflow and Data Integration Diagrams
Nuclei Isolation and Benchmarking Workflow
Integrated Data Analysis Logic
Comparing Enzymatic vs. Mechanical Dissociation Artefacts
The isolation of high-quality nuclei from frozen postmortem brain tissue is a critical prerequisite for single-nucleus RNA sequencing (snRNA-seq), a powerful tool for investigating cellular heterogeneity in complex neurological tissues and drug development research. The choice between enzymatic dissociation and mechanical dissociation represents a fundamental methodological crossroads, with each approach introducing distinct artefacts that can profoundly impact data integrity and biological interpretation [3] [48]. Enzymatic methods, while often providing higher yields, induce significant transcriptional stress responses and proteotype alterations in sensitive neural cells like microglia and astrocytes [48] [49]. Mechanical approaches better preserve in vivo transcriptional states but may result in lower nuclei yields and require careful optimization to prevent nuclear damage [3] [50]. This Application Note systematically compares these artefacts within the context of frozen postmortem brain tissue research and provides optimized protocols to minimize technical confounders.
Quantitative Comparison of Dissociation Artefacts
Performance Metrics Across Isolation Methods
Table 1: Comparative performance of nuclei isolation methods for brain tissue
| Performance Metric | Sucrose Gradient Centrifugation | Spin Column-Based Method | Machine-Assisted Platform |
|---|---|---|---|
| Nuclei Yield (per mg tissue) | ~60,000 | ~25% lower than other methods | ~60,000 |
| Structurally Intact Nuclei | 85% | 35% | ~100% |
| Debris Contamination | Minimal | Substantial | Negligible |
| Astrocyte Proportion | 13.9% | Intermediate | Lower |
| Microglia Proportion | Lower | Intermediate | 5.6% |
| Oligodendrocyte Proportion | Lower | Intermediate | 15.9% |
| Transcriptional Homogeneity (ROGUE) | Moderate | Lower | Moderate to High |
Data adapted from systematic comparison of three mechanistically distinct nuclei isolation strategies using mouse brain cortex [3].
Cell-Type-Specific Artefacts and Yield
Table 2: Cell-type-specific artefacts and yields observed with different dissociation methods
| Cell Type | Enzymatic Dissociation Artefacts | Mechanical Dissociation Advantages | Key Affected Markers |
|---|---|---|---|
| Microglia | Profound activation; 226 differentially expressed genes; distinct ex vivo activated population | Preserved homeostatic state; minimal stress gene induction | ↑ Fos, Jun, Hspa1a, Ccl3, Ccl4 (ENZ) ↑ Tmem119, P2ry12, CX3CR1 (MECH) |
| Astrocytes | 290 differentially expressed genes; altered metabolic protein profiles | Maintained in vivo proteotype; reduced stress responses | ↑ Gja1, Slc1a3 (both methods) |
| Neurons | 771 differentially expressed genes; highest sensitivity to enzymatic treatment | Better preservation of neuronal subtypes; reduced IEG expression | ↑ Sv2b, Slc17a7 (excitatory) ↑ Gad1, Gad2 (inhibitory) |
| Oligodendrocytes | 369 differentially expressed genes | Improved lineage representation | - |
Data compiled from multiple studies investigating transcriptional and proteotype responses to dissociation methods [48] [49].
Experimental Protocols
Optimized Mechanical Dissociation Protocol for Frozen Postmortem Brain Tissue
Principle: This protocol utilizes cold mechanical forces to isolate nuclei while maintaining transcriptional fidelity, minimizing ex vivo stress responses in sensitive neural cells [48] [49].
Reagents:
- Homogenization Buffer: 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl₂, 0.05% NP-40, 0.25% Glycerol, 5% BSA, 40 U/mL RNase inhibitor [5]
- Nuclei Washing Buffer: 0.5X PBS, 5% BSA, 0.25% Glycerol, 40 U/mL RNase inhibitor [5]
- Iodixanol (Optiprep) Solution: 29% (wt/vol) in nuclei washing buffer [5]
Procedure:
- Tissue Preparation: Maintain frozen brain tissue (25-50 mg) on dry ice throughout weighing and transfer to pre-chilled 1.5 mL tube [2].
- Homogenization: Add 3 mL ice-cold homogenization buffer with NP-40. Dounce homogenize with 15-20 strokes using tight clearance pestle (clearance: 0.0005-0.0025 inches) [5].
- Lysis Incubation: Incubate homogenate on ice for 5 minutes [5].
- Filtration: Pass suspension through 30 μm MACS strainers to remove debris [5].
- Centrifugation: Centrifuge at 1000 g for 10 minutes at 4°C [5].
- Density Gradient: Resuspend pellet in 1 mL nuclei washing buffer, mix with 1 mL 50% iodixanol, layer over 2 mL cushion of 29% iodixanol [5].
- Centrifugation & Collection: Centrifuge at 1000 g for 10 minutes at 4°C. Collect nuclei at interface [5].
- Wash & Resuspend: Wash nuclei with 1 mL nuclei washing buffer, centrifuge, and resuspend in appropriate buffer for downstream applications [5].
Critical Considerations:
- Maintain cold conditions (4°C) throughout procedure [48]
- Optimize homogenization strokes for specific tissue type (fewer for neuronal, more for fibrous tissue) [5]
- Include RNase inhibitors at all steps to preserve RNA integrity [2]
Inhibitor-Modified Enzymatic Dissociation Protocol
Principle: This protocol incorporates transcriptional and translational inhibitors during enzymatic digestion to mitigate dissociation-induced artefacts while maintaining higher nuclei yields [49].
Reagents:
- Enzymatic Digestion Solution: Trypsin-EDTA (0.125%), Collagenase, DNase, BSA in PBS [51]
- Inhibitor Cocktail: Actinomycin D (transcriptional inhibitor), Cycloheximide (translational inhibitor) [49]
- Blocking Buffer: PBS with 5% BSA [2]
Procedure:
- Tissue Preparation: Mince 25-50 mg frozen brain tissue into ~1 mm³ pieces on dry ice [2].
- Inhibitor Pre-treatment: Incubate tissue with inhibitor cocktail on ice for 15 minutes [49].
- Enzymatic Digestion: Add enzymatic digestion solution with maintained inhibitors. Digest at 37°C for 30-60 minutes with gentle agitation [49].
- Reaction Termination: Add excess cold blocking buffer [2].
- Mechanical Dissociation: Gently triturate with wide-bore pipette tips (5-10 strokes) [50].
- Filtration: Filter through 40 μm cell strainers [52].
- Centrifugation & Wash: Centrifuge at 500 g for 5 minutes at 4°C. Wash pellet with cold blocking buffer [2].
- Debris Removal: Use density gradient centrifugation as in mechanical protocol if debris is substantial [5].
Critical Considerations:
- Include inhibitor-only controls to assess off-target effects [49]
- Titrate enzyme concentrations to minimum effective level [50]
- Limit enzymatic digestion time to minimum required for tissue dissociation [53]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key reagents and their functions in nuclei isolation protocols
| Reagent/Category | Function | Example Applications |
|---|---|---|
| Detergents | Membrane permeabilization, nuclear release | NP-40 (0.05-0.1%): controlled lysis [5] |
| Enzymes | Extracellular matrix digestion, tissue dissociation | Trypsin, Collagenase: enzymatic dissociation [51] [50] |
| RNase Inhibitors | RNA integrity preservation | Protector RNase Inhibitor: throughout isolation [5] |
| Density Gradient Media | Debris removal, nuclei purification | Iodixanol (Optiprep): nuclei purification [5] |
| Inhibitors | Prevention of ex vivo gene expression | Actinomycin D: blocks transcription [49] |
| Antibodies | Cell type-specific nuclei isolation | NeuN: neuronal nuclei enrichment [2] |
| Buffering Systems | pH maintenance, osmotic balance | Tris-HCl, PBS: stable ionic environment [5] |
| Fluorescent Stains | Viability assessment, nuclei quantification | DAPI, Propidium Iodide: nuclei identification [2] |
Workflow and Pathway Diagrams
Diagram 1: Dissociation methods and transcriptional artefacts. This workflow illustrates the distinct artefact profiles and downstream impacts of enzymatic versus mechanical dissociation methods on snRNA-seq data from brain tissue.
Diagram 2: Unified nuclei isolation workflow. This optimized protocol integrates critical steps from both mechanical and enzymatic approaches, highlighting temperature control and minimal processing time as essential factors for success.
The selection between enzymatic and mechanical dissociation methods for nuclei isolation from frozen postmortem brain tissue involves careful consideration of competing priorities. Enzymatic approaches can provide higher nuclei yields but introduce significant transcriptional artefacts that confound biological interpretation, particularly in stress-sensitive cells like microglia. Mechanical methods better preserve native transcriptional states but require optimization to maintain yield and representativeness. The integration of transcriptional inhibitors during enzymatic dissociation or the adoption of optimized cold mechanical protocols presented herein provides researchers with validated strategies to minimize technical artefacts. As single-nucleus technologies continue to advance toward clinical applications in drug development, stringent standardization of dissociation methodologies will be paramount for generating biologically meaningful data from precious frozen postmortem brain specimens.
The critical assessment of nuclei count, viability, and doublet rates forms the essential quality control triad for successful single-nuclei genomics in frozen postmortem brain research. These technical performance metrics directly determine the feasibility and reliability of downstream omics applications, from single-nuclei RNA sequencing to epigenomic profiling. Within the broader context of optimizing nuclei isolation protocols from frozen postmortem brain tissue, rigorous quantification of these parameters ensures that resulting data accurately reflect biological reality rather than technical artifacts. This application note provides a structured framework for evaluating these key performance indicators, supported by experimental data and detailed methodologies essential for researchers, scientists, and drug development professionals working with precious brain bank specimens.
Quantitative Performance Metrics
The following tables consolidate key quantitative metrics from optimized nuclei isolation protocols for frozen postmortem brain tissue, providing reference values for technical performance assessment.
Table 1: Technical Performance Metrics for Nuclei Isolation from Frozen Postmortem Brain Tissue
| Parameter | Typical Range | Measurement Method | Significance for Downstream Applications |
|---|---|---|---|
| Nuclei Count (Yield) | Varies by input; ~5 million nuclei from 1000 mg tissue [54] | Automated cell counters (e.g., Countess 3 FL); staining with Acridine Orange/Propidium Iodide [2] | Determines feasibility of sequencing assays and required sequencing depth |
| Nuclei Viability/Integrity | Majority double-stained (AO/PI) or RFP-labeled [2] | Fluorescent viability stains (e.g., ReadyCount Red/Green); microscopy [2] [5] | Induces background noise in sequencing; low viability suggests protocol degradation |
| Doublet Rate | PCR duplication rate ~13.8% in methyl-seq [2] | Computational detection (e.g., DoubletFinder in snRNA-seq) [5] | Can confound biological interpretation by creating artificial cell types |
Table 2: Key Reagents for Nuclei Isolation and Quality Control
| Reagent / Kit | Specific Example | Function in Protocol |
|---|---|---|
| Nuclei Isolation Kit | 10X Genomics Chromium Nuclei Isolation Kit [2] | Provides optimized buffers for tissue lysis and nuclei stabilization |
| Viability Stain | Invitrogen ReadyCount Red/Green (AO/PI) [2] | Differential staining of intact vs. compromised nuclei |
| Nuclear Stain | DAPI (4',6-diamidino-2-phenylindole) [8] | DNA stain for flow cytometry gating and total nuclei identification |
| Antibody for Sorting | Anti-NeuN (Neuronal nuclei) [2] [8] [54] | Immunostaining for fluorescence-activated nuclei sorting (FANS) of neurons |
| DNA Extraction Kit | QIAamp DNA Micro Kit [2] | Nucleic acid extraction optimized for low-input samples post-sorting |
| RNase Inhibitor | Protector RNase Inhibitor [5] | Preserves RNA integrity during nuclei isolation for snRNA-seq |
Experimental Protocols for Technical Assessment
Nuclei Quantification and Viability Staining Protocol
Principle: This protocol enables accurate enumeration of nuclei yield and assessment of integrity using membrane-permeant (Acridine Orange) and membrane-impermeant (Propidium Iodide) fluorescent nucleic acid stains [2].
Procedure:
- Following nuclei isolation, combine 5 µL of nuclei suspension with 5 µL of ReadyCount Red/Green viability stain.
- Pipette the mixture up and down to ensure homogeneous staining.
- Load the stained suspension into a counting chamber slide for analysis on an automated cell counter (e.g., Countess 3 FL).
- Using fluorescent cubes for GFP (Acridine Orange) and RFP (Propidium Iodide), quantify the nuclei populations.
- Interpretation: A successful isolation shows a majority of nuclei stained with both dyes or with Propidium Iodide alone, indicating intact nuclei with permeable outer membranes [2].
Flow Cytometry and Fluorescence-Activated Nuclei Sorting (FANS)
Principle: This method separates nuclei based on cell-type-specific markers (e.g., NeuN for neurons) and size, enabling collection of pure populations for downstream applications while providing data on sample quality [2] [8].
Procedure:
- Immunostaining: Incubate nuclei suspension with primary antibody (e.g., NeuN) for 30 minutes on ice. Include unstained and secondary-antibody-only controls [2].
- Wash and Secondary Staining: Pellet nuclei by centrifugation (400 rcf, 5 min), remove supernatant, and resuspend in blocking buffer. Add fluorescently-labeled secondary antibody and incubate 15 minutes in the dark on ice [2].
- DNA Staining: Prior to sorting, add DAPI (1 µL of 0.1 mg/mL) to identify total nuclei population [8].
- Gating Strategy:
- First, gate on DAPI-positive events to exclude non-nuclear debris.
- Next, apply a singlet gate based on forward scatter area vs. height to exclude doublets and aggregates.
- Finally, set fluorescence gates based on the secondary antibody channel, using the controls to establish a negative threshold and identify positive populations (e.g., NeuN+ neurons) [2] [54].
- Sorting: Collect purified nuclei populations into tubes containing chilled blocking buffer. Validate sort purity by re-running a sorted sample [2].
Computational Doublet Detection in Single-Nuclei RNA-seq Data
Principle: Bioinformatic tools identify droplets containing more than one nucleus by detecting unique gene expression patterns that represent averaged transcriptional profiles of multiple cell types [5].
Procedure:
- Process sequencing data through standard snRNA-seq pipelines (e.g., Cell Ranger) to generate gene expression matrices.
- Import filtered data into R/Python environments and apply doublet detection algorithms (e.g., DoubletFinder).
- Set expected doublet rates based on the number of nuclei loaded, as higher loading concentrations increase doublet formation probability.
- Remove identified doublets from the dataset before proceeding with downstream analysis such as clustering and differential expression.
- Quality Metric: Report the percentage of identified doublets as a key quality control metric for the experiment [5].
Workflow and Logical Relationships
The following diagram illustrates the complete workflow for nuclei isolation from frozen postmortem brain tissue and the associated quality control checkpoints to assess technical performance.
Nuclei Isolation and Quality Control Workflow
Discussion
The quantitative assessment of nuclei count, viability, and doublet rates provides critical insights into the success of isolation protocols and predicts downstream analytical performance. When nuclei yields fall below expectations, researchers should consider modifying lysis conditions, increasing filtration steps, or adding wash steps to reduce debris [2]. For challenging postmortem primate brain tissue with high myelin content, incorporating a sucrose cushion or iodixanol gradient during centrifugation can significantly improve purity by effectively separating nuclei from lipid-rich debris [8] [54].
Low viability metrics often reflect suboptimal tissue preservation or excessive mechanical stress during homogenization. While the postmortem interval inevitably affects tissue integrity, adjusting dounce strokes, using looser-fitting pestles, and maintaining consistent cold temperatures during processing can help preserve nuclear integrity [5]. Elevated doublet rates typically originate from overcrowding during sorting or sequencing library preparation. Accurate nuclei concentration quantification and careful calibration of loading concentrations are essential mitigation strategies.
The integration of these technical performance assessments creates a robust framework for optimizing nuclei isolation from frozen postmortem brain tissue. By systematically tracking these metrics across experiments, researchers can establish laboratory-specific benchmarks, identify procedural inconsistencies, and ultimately generate higher quality genomic and epigenomic data from these invaluable clinical specimens.
Single-nucleus RNA sequencing (snRNA-seq) has emerged as a transformative technology for investigating complex tissues, such as the brain, where traditional single-cell approaches are hampered by the inability to obtain fresh samples and the large cell size of key populations like neurons. This application note details a successful case study integrating an optimized nuclei isolation protocol from frozen postmortem brain tissue with downstream snRNA-seq workflows. The protocol is specifically designed to address the unique challenges presented by nonhuman primate (NHP) brain tissue, including high levels of myelin debris and reduced RNA integrity, which are common in precious samples from our closest living relatives [2]. The methodologies and findings presented herein are framed within a broader thesis on advancing nuclei isolation techniques for brain research, providing a validated roadmap for researchers and drug development professionals aiming to leverage archived biospecimens for high-resolution transcriptomic studies.
Methodologies: Optimized Nuclei Isolation & Workflow Integration
Optimized Nuclei Isolation from Frozen Postmortem Brain Tissue
The foundational step for successful snRNA-seq integration is the efficient isolation of high-quality nuclei. The following protocol was optimized for ~25 mg of frozen postmortem chimpanzee cerebral cortex tissue and is highly applicable to other NHP or human brain samples [2].
- Tissue Dissection: Hemisectioned whole brain slabs stored at -80°C were microdissected on dry ice using a 2 mm biopsy punch, yielding ~10-25 mg tissue pieces. Approximately 25-50 mg of frozen tissue was weighed per isolation reaction in pre-chilled 1.5 mL microcentrifuge tubes [2].
- Nuclei Isolation and Lysis: The 10X Genomics Chromium Nuclei Isolation Kit was used as a base, with critical optimizations for brain tissue. Key modifications included:
- Adapted Lysis Conditions: Lysis time was carefully optimized to balance complete tissue disruption with the preservation of nuclear membrane integrity.
- Enhanced Filtration: A sequential filtration step was introduced using a Flowmi cell strainer (40 µm) to remove large debris and myelin aggregates.
- Additional Wash Steps: Extra washes were incorporated to reduce background debris and soluble contaminants, which are particularly prevalent in postmortem brain tissue [2].
- Nuclei Quantification and Quality Control: The nuclei suspension was quantified using an automated cell counter (e.g., Countess 3 FL). Viability stains Acridine Orange (AO) and Propidium Iodide (PI) were used; a successful isolation is indicated by a majority of nuclei being stained with the membrane-impermeable PI, confirming intact nuclei [2].
- Optional Fluorescent-Activated Nuclei Sorting (FANS): For cell type-specific applications, an immunostaining step can be integrated.
- Staining: Nuclei were stained with a primary antibody against the neuronal marker NeuN (Hexaribonucleotide Binding Protein-3) for 30 minutes on ice, followed by a secondary antibody incubation for 15 minutes in the dark [2].
- Sorting: Stained nuclei were sorted on a flow cytometer (e.g., Sony SH800Z). Gates were established using unstained and secondary antibody-only controls, with DAPI used to identify intact nuclei. This allows for the enrichment of neuronal (NeuN+) and non-neuronal (NeuN-) populations for bulk or single-nucleus analyses [2].
Integrated Single-Nuclei Multiome Sequencing Workflow
For a comprehensive profile, the isolated nuclei can be directly input into multiome sequencing workflows. A generalized, robust protocol for single-nucleus multiome sequencing (snMultiome-seq) from frozen tissues involves the following steps, which can be applied to brain nuclei [55]:
- Library Construction: Isolated nuclei are loaded onto a 10X Genomics Chromium chip to create Gel Bead-In-Emulsions (GEMs). Within these GEMs, both nuclear transcriptomes (snRNA-seq) and chromatin accessibility (snATAC-seq) are barcoded simultaneously from the same nucleus [55].
- Sequencing: The resulting libraries are sequenced on a high-throughput platform (e.g., Illumina NovaSeq) to a sufficient depth (e.g., >20,000 paired-end reads per nucleus) [5].
- Data Processing and Integration:
- Primary Analysis: Raw sequencing data (FastQ files) are processed through standard pipelines (e.g., Cell Ranger) for alignment, barcode assignment, and count matrix generation [5].
- Quality Control: Nuclei are filtered based on metrics like total UMI counts (nCountRNA), number of detected genes (nFeatureRNA), and mitochondrial gene percentage. Tools like DoubletFinder can be used to remove doublets [5].
- Modality Integration: A key challenge is integrating snRNA-seq data with other modalities, such as single-cell RNA-seq (scRNA-seq) from fresh tissue. Effective strategies include:
- Gene Filtering: Pruning genes that show consistent differential expression between snRNA-seq and scRNA-seq data significantly improves the accuracy of integrated analyses and bulk deconvolution [56].
- Harmonization Algorithms: Using conditional variational autoencoders (e.g., conditional scVI) can effectively harmonize data across modalities, especially when matched cell-type references are unavailable [56].
Workflow Visualization
The following diagram illustrates the complete integrated workflow, from tissue to data analysis:
Workflow from Tissue to Data Analysis
A second diagram outlines the core decision-making process for integrating single-nucleus and single-cell data:
Data Integration Strategy Decision Tree
Performance of the Optimized Nuclei Isolation Protocol
The table below summarizes the quantitative outcomes of applying the optimized nuclei isolation protocol to frozen postmortem chimpanzee brain tissue, as validated by downstream applications [2].
Table 1: Nuclei Isolation and Sequencing Outcomes from Chimpanzee Cortex
| Parameter | Result / Value | Downstream Application & Outcome |
|---|---|---|
| Starting Tissue Input | ~25 mg | Enables analysis of small, region-specific biopsies. |
| Key Protocol Steps | Adapted lysis, enhanced filtration, additional washes | Reduced debris and improved nuclei integrity for flow cytometry. |
| NeuN+ FANS Yield | Successful enrichment of neuronal nuclei | Compatible with bulk methylome sequencing of neuronal populations. |
| Methyl-seq Coverage | >7X coverage of genome-wide CpGs | Achieved using gDNA from FANS-sorted neuronal nuclei; PCR duplication rate of 13.8%. |
| Library Prep Kit | NEBNext Enzymatic Methyl-seq Kit | Recommended for low DNA input due to less DNA damage compared to bisulfite conversion. |
Application Across Diverse Tissue Types
The versatility of a similar low-input nuclei isolation approach is demonstrated by its application to various human cancer tissues, yielding data comparable to established single-cell atlases [5].
Table 2: snRNA-seq Results from Low-Input Cryopreserved Human Tissues
| Tissue Type | Number of Nuclei Profiled (Post-QC) | Key Quality Control Metrics |
|---|---|---|
| Brain (Cancer) | 1,550 - 7,468 nuclei | nCountRNA ≥ 300, nFeatureRNA 250-2500, mitochondrial percentage < 10% [5]. |
| Bladder (Cancer) | 1,550 - 7,468 nuclei | nCountRNA ≥ 300, nFeatureRNA 250-2500, mitochondrial percentage < 10% [5]. |
| Lung (Cancer) | 1,550 - 7,468 nuclei | nCountRNA ≥ 300, nFeatureRNA 250-2500, mitochondrial percentage < 10% [5]. |
| Prostate (Cancer) | 1,550 - 7,468 nuclei | nCountRNA ≥ 300, nFeatureRNA 250-2500, mitochondrial percentage < 10% [5]. |
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and kits critical for replicating the successful snRNA-seq integration workflow described in this case study.
Table 3: Essential Reagents and Kits for snRNA-seq Workflows
| Item | Function / Application | Specific Example / Note |
|---|---|---|
| Nuclei Isolation Kit | Base protocol for extracting nuclei from frozen tissue. | 10X Genomics Chromium Nuclei Isolation Kit; requires optimization for brain tissue [2]. |
| Dounce Homogenizer | Mechanical tissue dissociation. | Used with loose and tight pestles; optimal strokes vary by tissue [5]. |
| Flowmi Cell Strainer | Removal of large debris and myelin aggregates. | 40 µm size used for post-lysis filtration [2]. |
| Anti-NeuN Antibody | Immunostaining for neuronal nuclei enrichment via FANS. | Primary antibody for flow sorting of neuronal populations [2]. |
| Fluorescent Viability Stain | Quantification of nuclei integrity and concentration. | e.g., ReadyCount Red/Green (contains AO/PI); PI stains nuclei after lysis [2]. |
| 10X Genomics Multiome Kit | Simultaneous snRNA-seq and snATAC-seq library generation. | For integrated transcriptomic and epigenomic profiling from the same nucleus [55]. |
| IODIXANOL (Optiprep) | Density gradient medium for nuclei purification. | Used in density gradient centrifugation to purify nuclei from debris [5]. |
| RNase Inhibitor | Preservation of RNA integrity during isolation. | Critical for maintaining mRNA quality in postmortem samples [2]. |
This case study demonstrates a robust and integrated workflow for nuclei isolation from challenging frozen postmortem brain tissue and its successful application in snRNA-seq. The optimized protocol directly addresses the major hurdles of low input mass, high debris, and variable RNA integrity, enabling the study of rare and valuable NHP specimens [2]. The integration with FANS further expands its utility by allowing for cell type-specific epigenomic analyses, such as methylome sequencing, providing a deeper layer of biological insight.
The successful application of a analogous low-input protocol across diverse cancer tissues underscores its versatility and robustness for profiling cryopreserved biospecimens [5]. Furthermore, the clear guidelines for data integration—prioritizing scRNA-seq references with snRNA-seq appended after filtering cross-modality DEGs, or using conditional scVI for less-characterized systems—provide a solid bioinformatic foundation for combining datasets across modalities [56]. For researchers in neuroscience and drug development, this end-to-end workflow offers a reliable path to unlock the potential of archived frozen brain tissues, accelerating discovery in brain function, evolution, and disease.
Conclusion
The isolation of high-quality nuclei from frozen postmortem brain tissue is a powerful gateway to unlocking the cellular and molecular complexity of the brain using modern genomic tools. By understanding the foundational rationale, meticulously applying optimized protocols, proactively troubleshooting, and rigorously validating outputs, researchers can reliably generate robust data from even rare and low-input biobanked samples. This capability is pivotal for advancing our understanding of neurodevelopment, aging, and the pathophysiology of neurological and psychiatric disorders, ultimately accelerating the discovery of novel therapeutic targets. Future directions will focus on further miniaturization of protocols, integration of multi-omic assays from the same nuclei, and the development of standardized cross-laboratory pipelines to enhance reproducibility and data sharing in neuroscience.
References
- 1. Mapping human brain cell type origin and diseases ... [https://www.nature.com/articles/s41398-025-03562-6]
- 2. For All the Primate FANS: Optimized Isolation of Nuclei ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204041/]
- 3. Comparative analysis of nuclei isolation methods for brain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11974938/]
- 4. A single-cell and single-nucleus RNA-Seq toolbox for fresh ... [https://www.nature.com/articles/s41591-020-0844-1]
- 5. A versatile and efficient method to isolate nuclei from low- ... [https://www.nature.com/articles/s41598-025-90070-8]
- 6. Isolation of nuclei from frozen brain tissue. [https://bio-protocol.org/exchange/minidetail?id=16077445&type=30]
- 7. Multiplexed Fixed Single Cell RNA Sequencing of frozen ... [https://www.protocols.io/view/multiplexed-fixed-single-cell-rna-sequencing-of-fr-dmig44bw.html]
- 8. Nuclei isolation of multiple brain cell types for omics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7969463/]
- 9. scRNAseq vs snRNAseq [https://levitasbio.com/scrnaseq-vs-snrnaseq/]
- 10. Nuclei Isolation from Embryonic Mouse Brain Tissue for ... [https://www.10xgenomics.com/support/epi-multiome/documentation/steps/sample-prep/nuclei-isolation-from-embryonic-mouse-brain-tissue-for-single-cell-multiome-atac-plus-gene-expression-sequencing]
- 11. Multimodal single- cell and whole -genome sequencing of small, frozen ... [https://pubmed.ncbi.nlm.nih.gov/36624340/]
- 12. A simplified preparation method for single-nucleus RNA ... [https://www.nature.com/articles/s41598-025-97053-9]
- 13. Exploring Key Applications of Single Nuclei RNA Sequencing [https://www.denovix.com/exploring-key-applications-of-single-nuclei-rna-sequencing]
- 14. Clinical epigenetics: recent advances and opportunities [https://epicom.biomedcentral.com/articles/10.1186/s43682-025-00038-y]
- 15. Protocol for achieving enhanced snRNA-seq data quality ... [https://www.sciencedirect.com/science/article/pii/S2666166725001236]
- 16. Cryopreservation of brain cell structure: a review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11753176/]
- 17. An optimized protocol for single nuclei isolation from ... [https://www.nature.com/articles/s41598-022-14099-9]
- 18. Cryoprotectant Protocols for Storage of Free-Floating Brain ... [https://ihcworld.com/2024/01/25/cryoprotectant-protocols-for-storage-of-free-floating-brain-sections/]
- 19. Nuclei isolation from brain tissue for single-cell ATAC [https://www.protocols.io/view/nuclei-isolation-from-brain-tissue-for-single-cell-g4ctbyswp.html]
- 20. Using mechanical homogenization to isolate microglia from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9485281/]
- 21. Gentle Isolation of Nuclei from Brain Tissue of Adult African ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9828740/]
- 22. A method for rapid flow-cytometric isolation of endothelial ... [https://www.nature.com/articles/s41374-021-00698-z]
- 23. Nuclei isolation for single-nuclei rna sequencing - Technical [https://community.brain-map.org/t/nuclei-isolation-for-single-nuclei-rna-sequencing/795]
- 24. Nuclei isolation from fresh frozen tissues for 10X Single ... [https://www.protocols.io/view/nuclei-isolation-from-fresh-frozen-tissues-for-10x-dxey7jfw.html]
- 25. How to Isolate Nuclei for Single Cell Methods [https://singleron.bio/how-to-isolate-nuclei-for-single-cell-methods/]
- 26. Systematic Methods for Isolating High Purity Nuclei from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9740283/]
- 27. Optimizing high-throughput viral vector characterization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10444642/]
- 28. NeuN As a Neuronal Nuclear Antigen and ... - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4463411/]
- 29. NeuN [https://en.wikipedia.org/wiki/NeuN]
- 30. Anti-NeuN antibody - Neuronal Marker (ab134014) [https://www.abcam.com/en-us/products/primary-antibodies/neun-antibody-neuronal-marker-ab134014]
- 31. Optimized nucleus isolation protocol from frozen mouse ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2023.1243863/full]
- 32. Nuclei isolation, immunostaining, and Fluorescence ... [https://www.protocols.io/view/nuclei-isolation-immunostaining-and-fluorescence-a-ddz52786.html]
- 33. Isolation of nuclei from fresh frozen brain sections V.1 [https://www.protocols.io/view/isolation-of-nuclei-from-fresh-frozen-brain-sectio-dys97wh6.html]
- 34. A Simple, Quick, and Partially Automated Protocol for the ... [https://www.jove.com/t/65611/a-simple-quick-partially-automated-protocol-for-isolation-single]
- 35. Postmortem Interval Leads to Loss of Disease-Specific ... [https://www.eneuro.org/content/12/3/ENEURO.0505-24.2025]
- 36. RNA Quality in Post-mortem Human Brain Tissue Is ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2021.780352/full]
- 37. A method for rapid flow-cytometric isolation of endothelial ... [https://www.sciencedirect.com/science/article/pii/S0023683722000824]
- 38. Optimized Isolation of Nuclei from Frozen Postmortem Primate ... [https://scholars.duke.edu/publication/1679844]
- 39. How to Quantify Isolated Nuclei from Brain Tissue [https://www.denovix.com/blog/how-to-quantify-isolated-nuclei-from-brain-tissue/]
- 40. Flow Cytometry: An Overview - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5939936/]
- 41. Considerations for Flow Cytometry Gating | FACS Analysis [https://www.stemcell.com/considerations-for-facs-gating.html]
- 42. Gating [https://docs.cellengine.com/gate/]
- 43. Cytometry in the brain: studying differentiation to diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6496381/]
- 44. Use of flow cytometry for high-throughput cell population ... [https://www.frontiersin.org/journals/neuroanatomy/articles/10.3389/fnana.2012.00027/full]
- 45. CelltypeR: A flow cytometry pipeline to characterize single ... [https://www.sciencedirect.com/science/article/pii/S2589004224018388]
- 46. An integrated single-nucleus and spatial transcriptomics ... [https://www.nature.com/articles/s41593-025-02022-0]
- 47. An Efficient Method for Isolating and Purifying Nuclei from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10855070/]
- 48. Enzymatic Dissociation Induces Transcriptional and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7663484/]
- 49. Dissection of artifactual and confounding glial signatures ... [https://www.nature.com/articles/s41593-022-01022-8]
- 50. Tissue dissociation for single-cell and single-nuclei RNA ... [https://frontiersinzoology.biomedcentral.com/articles/10.1186/s12983-022-00472-x]
- 51. Automated—Mechanical Procedure Compared to Gentle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9138555/]
- 52. Optimized nucleus isolation protocol from frozen mouse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10575574/]
- 53. snCED-seq: high-fidelity cryogenic enzymatic dissociation ... [https://www.nature.com/articles/s41467-025-59464-0]
- 54. Neuronal Nuclei from Human Isolation Postmortem Brain Tissue [https://www.jove.com/t/914/neuronal-nuclei-isolation-from-human-postmortem-brain-tissue]
- 55. A simple approach of nuclei isolation for single nucleus ... [https://www.sciencedirect.com/science/article/abs/pii/S0022282825001506]
- 56. Integrating single-cell and single-nucleus datasets ... [https://pubmed.ncbi.nlm.nih.gov/40894744/]