Locus-Specific vs. Genome-Wide DNA Methylation Analysis: A Strategic Guide for Biomedical Researchers
Selecting the appropriate DNA methylation analysis method is a critical strategic decision that directly impacts the success and cost-effectiveness of epigenetic research and biomarker development.
Locus-Specific vs. Genome-Wide DNA Methylation Analysis: A Strategic Guide for Biomedical Researchers
Abstract
Selecting the appropriate DNA methylation analysis method is a critical strategic decision that directly impacts the success and cost-effectiveness of epigenetic research and biomarker development. This article provides a comprehensive comparison of locus-specific and genome-wide methods, tailored for researchers and drug development professionals. We explore the foundational principles of DNA methylation, detail the methodologies and applications of key techniques—from bisulfite pyrosequencing and MS-HRM to whole-genome bisulfite sequencing (WGBS), microarrays, and emerging long-read technologies. A practical troubleshooting guide addresses common challenges like sample quality and data analysis. Finally, we present a rigorous validation framework and a comparative analysis to empower scientists in selecting the optimal methodology for their specific research goals, from targeted biomarker validation to novel epigenome discovery.
Core Principles of DNA Methylation and Analysis Strategies
5-Methylcytosine (5mC) represents a fundamental epigenetic modification where a methyl group is covalently bound to the fifth carbon of a cytosine base in DNA, profoundly influencing gene expression without altering the underlying DNA sequence [1]. This modification regulates gene transcription and serves critical biological roles across diverse organisms, from bacteria to mammals [1]. In vertebrates, 5mC predominantly occurs at CpG dinucleotides, with approximately 70-80% of CpG cytosines methylated, and clusters of CpG sites at the 5' ends of genes termed CpG islands [1]. The establishment, maintenance, and interpretation of 5mC patterns are essential for normal development, cellular differentiation, and disease pathogenesis, particularly in cancer and neurological disorders [1] [2].
The field of epigenetics has increasingly recognized that defining the precise genomic distribution of 5mC is crucial for understanding its biological functions. This has driven the development of numerous analytical technologies to map 5mC patterns at varying resolutions, from single loci to entire genomes [3] [4]. These methods largely fall into two categories: locus-specific approaches that interrogate predefined genomic regions, and genome-wide techniques that provide comprehensive methylome maps [3]. This review objectively compares the performance of these methodological approaches, providing experimental data and protocols to guide researchers in selecting appropriate strategies for investigating the biological roles of 5-methylcytosine.
Biological Foundations of 5-Methylcytosine
Establishment and Maintenance of DNA Methylation
In mammalian systems, 5mC marks are established and maintained by a family of DNA methyltransferases (DNMTs). Humans possess five DNMTs: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L [1]. DNMT3A and DNMT3B primarily catalyze de novo methylation, introducing new 5mC marks during embryonic development, while DNMT1 maintains these marks following DNA replication, copying methylation patterns from the parent strand to the daughter strand [1]. The catalytic mechanism involves a cysteine residue in the DNMT's PCQ motif creating a nucleophilic attack at carbon 6 of the target cytosine, followed by transfer of a methyl group from S-adenosylmethionine to carbon 5, resulting in 5-methylcytosine [1].
Active and Passive DNA Demethylation Pathways
DNA methylation patterns are dynamic and can be reversed through both passive and active demethylation pathways. Passive demethylation occurs gradually through dilution when methylated cytosines are not maintained during DNA replication cycles [1]. In contrast, active DNA demethylation involves enzymatic processes where 5mC is iteratively oxidized by TET (ten-eleven translocation) family dioxygenases to 5-hydroxymethylcytosine (5hmC), then to 5-formylcytosine (5fC), and finally to 5-carboxylcytosine (5caC) [1]. The latter two oxidized derivatives are excised by thymine DNA glycosylase (TDG) and repaired via the base excision repair (BER) pathway to restore an unmethylated cytosine [1]. This active demethylation pathway requires Fe(II) as a cofactor and oxygen and α-ketoglutarate (α-KG) as substrates [1].
Functional Consequences in Transcription and Disease
The genomic context of 5mC significantly determines its functional impact. When located in gene promoter regions, particularly within CpG islands, 5mC typically associates with gene silencing by impeding transcription factor binding or recruiting proteins that promote chromatin condensation [1] [2]. In contrast, methylation within gene bodies correlates with active transcription, though the mechanistic basis for this association remains less clear [2]. Aberrant 5mC patterns represent a hallmark of various diseases, particularly cancer, where hypermethylation of tumor suppressor gene promoters can lead to their silencing, while hypomethylation of repeat sequences and oncogenes can promote genomic instability and aberrant activation [1] [5].
Figure 1: 5-Methylcytosine Metabolism Pathway. This diagram illustrates the establishment of 5mC by DNMTs using S-adenosylmethionine (SAM) as a methyl donor, and its stepwise oxidation and removal through the active demethylation pathway involving TET enzymes and base excision repair.
Comparative Analysis of DNA Methylation Detection Methods
Technology Categories and Principles
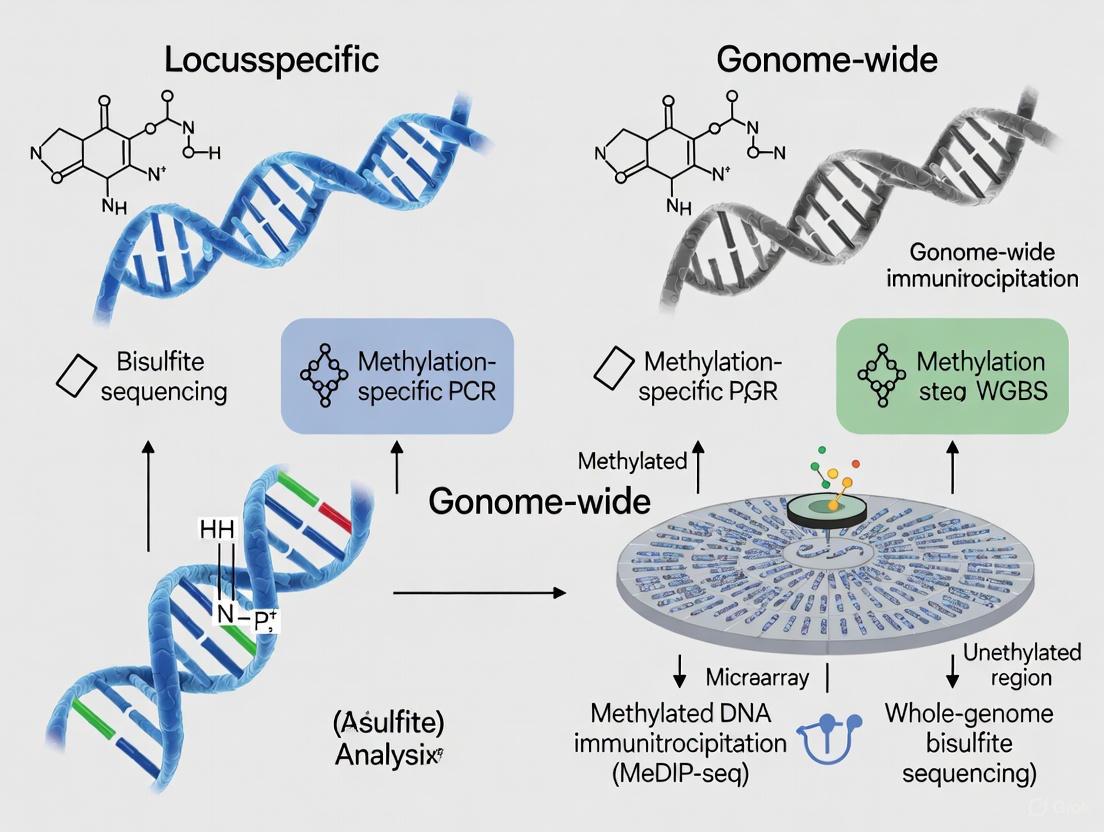
Methods for analyzing DNA methylation patterns rely on a limited number of core principles for differentiating methylation states: methylation-specific/methylation-dependent restriction enzymes, antibodies or methyl-binding proteins, chemical-based enrichment, and bisulfite conversion [3]. Bisulfite conversion represents the most widely adopted approach, particularly for clinical applications, as it converts unmethylated cytosines to uracils while leaving methylated cytosines unchanged, thereby creating sequence polymorphisms that can be detected through various downstream analysis platforms [1] [4].
DNA methylation assays can be broadly categorized into three groups based on their analytical scope and output:
Absolute DNA methylation assays provide quantitative measurements of DNA methylation levels at single-CpG resolution and include amplicon bisulfite sequencing (AmpliconBS), enrichment bisulfite sequencing (EnrichmentBS), mass spectrometric analysis (EpiTyper), and bisulfite pyrosequencing (Pyroseq) [4].
Relative DNA methylation assays measure DNA methylation by comparing samples to a reference, typically detecting methylated DNA fragments in an excess of unmethylated fragments. These include MethyLight, methylation-specific melting assays (MS-HRM, MS-MCA), and quantitative methylation-specific PCR (qMSP) [4].
Global DNA methylation assays measure a sample's total DNA methylation content using techniques like high-performance liquid chromatography with mass spectrometry (HPLC-MS), immunoquantification (Immunoquant), or bisulfite pyrosequencing of repetitive elements [4].
Locus-Specific Versus Genome-Wide Methodologies
The choice between locus-specific and genome-wide approaches represents a fundamental strategic decision in experimental design, with each offering distinct advantages and limitations:
Locus-specific methods focus on predetermined genomic regions of interest, typically employing techniques such as bisulfite pyrosequencing, Methylation-Specific PCR (MSP), or targeted bisulfite sequencing. These approaches offer higher sensitivity, lower cost per sample, and compatibility with clinical samples including formalin-fixed paraffin-embedded (FFPE) tissue, but provide limited information outside targeted regions [4] [6].
Genome-wide methods provide comprehensive methylome maps through techniques like whole-genome bisulfite sequencing (WGBS), reduced representation bisulfite sequencing (RRBS), or array-based platforms (Infinium MethylationEPIC). These approaches enable hypothesis-free discovery of novel differentially methylated regions but require higher DNA input, incur greater computational burdens, and have higher per-sample costs [3] [4].
Table 1: Performance Comparison of Major DNA Methylation Analysis Methods
| Method | Resolution | DNA Input | Cost per Sample | Throughput | Best Applications |
|---|---|---|---|---|---|
| AmpliconBS [4] | Single-base | Low | $$ | Medium | Targeted validation; clinical diagnostics |
| Bisulfite Pyrosequencing [4] | Single-base | Low | $ | High | Biomarker validation; clinical testing |
| Methylation Arrays [4] | CpG-site | Medium | $$ | Very High | Epigenome-wide association studies |
| WGBS [3] | Single-base | High | $$$$ | Low | Comprehensive methylome mapping |
| RRBS [3] | Single-base | Medium | $$$ | Medium | Cost-effective genome-wide coverage |
| T-LRS [6] | Single-base, haplotype-aware | Medium-High | $$$ | Medium | Imprinting disorders; structural variants |
Experimental Performance Benchmarking
A comprehensive community-wide benchmarking study compared the performance of widely used DNA methylation analysis methods compatible with routine clinical use [4]. Researchers evaluated 21 locus-specific assays across 27 predefined genomic regions, plus six global assays, using 32 standardized reference samples that mimicked typical clinical scenarios, including tumor-normal pairs, drug-treated cells, titration series, and FFPE samples [4].
The study revealed that most established methods provided high accuracy and robustness, with amplicon bisulfite sequencing and bisulfite pyrosequencing demonstrating the best all-round performance across multiple metrics [4]. These techniques showed excellent sensitivity on low-input samples and reliable discrimination between cell types, making them particularly suitable for biomarker validation and clinical diagnostics [4]. The benchmarking also highlighted that array-based methods like the Infinium HumanMethylation450K provided reliable genome-wide data but with limitations in detecting methylation at non-CpG contexts and regions with low probe coverage [4].
For specialized applications such as imprinting disorders, emerging technologies like targeted long-read sequencing (T-LRS) using nanopore technology offer distinct advantages by providing haplotype-aware methylation phasing across extended genomic regions without requiring bisulfite conversion [6]. A recent study established a T-LRS system targeting 78 differentially methylated regions (DMRs) and 22 genes related to imprinting disorders, achieving median read coverage exceeding 40× per DMR and enabling precise classification of DMRs based on parent-of-origin methylation patterns [6].
Detailed Experimental Protocols
Bisulfite Pyrosequencing for Locus-Specific Analysis
Bisulfite pyrosequencing represents one of the most robust and quantitative methods for locus-specific DNA methylation analysis, combining bisulfite conversion with sequencing-by-synthesis technology [4].
Protocol Steps:
- DNA Extraction and Quantification: Isolate genomic DNA using standard phenol-chloroform or column-based methods. Quantify DNA using fluorometric methods for accurate measurement.
- Bisulfite Conversion: Treat 500-1000 ng genomic DNA with sodium bisulfite using commercial kits (e.g., EZ DNA Methylation Kit from Zymo Research). Program: 98°C for 10 minutes, 64°C for 2.5 hours, then hold at 4°C.
- PCR Amplification: Design primers flanking the target CpG sites using specialized software (e.g., PyroMark Assay Design). Perform PCR with bisulfite-converted DNA using hot-start Taq polymerase to minimize nonspecific amplification.
- Pyrosequencing: Prepare single-stranded DNA from PCR products using the Pyrosequencing Vacuum Prep Workstation. Sequence using the PyroMark Q96 instrument with dispensation order optimized for target CpGs.
- Data Analysis: Quantify methylation percentage at each CpG using PyroMark Q96 software, which calculates the ratio of C/T incorporation at each position.
Quality Control Measures:
- Include unmethylated and fully methylated control DNA in each experiment
- Monitor bisulfite conversion efficiency by assessing non-CpG cytosine conversion
- Set quality score thresholds for sequence reads (>90% confidence)
- Perform technical replicates for critical samples
Targeted Long-Read Sequencing for Imprinting Disorders
Nanopore-based targeted long-read sequencing enables haplotype-phased methylation analysis across large genomic regions, providing unique insights into imprinting disorders [6].
Protocol Steps:
- High-Molecular-Weight DNA Extraction: Isolate genomic DNA from leukocytes using the Monarch HMW DNA Extraction Kit, preserving long DNA fragments.
- DNA Shearing and Size Selection: Shear 2-3 μg DNA to 10-15 kb fragments using g-TUBE (Covaris). Remove short fragments using the Short Read Eliminator Kit.
- Library Preparation and Adaptive Sampling: Prepare sequencing libraries using the DNA Ligation Sequencing Kit V14 (Oxford Nanopore Technologies). Implement adaptive sampling during sequencing to enrich for 36.5 Mb target regions covering 78 DMRs and 22 imprinting-disorder-related genes.
- Sequencing: Load libraries on MinION or PromethION flow cells (R10.4.1) and sequence using GridION Mk1 or PromethION 2 devices with MinKNOW software.
- Data Analysis: Basecall raw signals using Guppy or Dorado. Align reads to reference genome using minimap2. Calculate methylation index (MI) for each CpG using modified base calling algorithms (e.g., Megalodon). Phase reads by parental origin using SNPs.
Quality Control Measures:
- Assess DNA integrity number (DIN) >7.0 for HMW DNA
- Target >40 reads per DMR for statistical reliability
- Include control samples with known methylation patterns
- Validate findings with orthogonal methods in diagnostic settings
Figure 2: Locus-Specific Methylation Analysis Workflow. This diagram outlines the core experimental workflow for targeted DNA methylation analysis, highlighting the shared initial steps of bisulfite conversion and PCR amplification, followed by method-specific sequencing approaches.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for DNA Methylation Analysis
| Reagent/Material | Function | Example Products | Application Notes |
|---|---|---|---|
| Bisulfite Conversion Kits | Chemical conversion of unmethylated C to U | EZ DNA Methylation Kit (Zymo Research), Epitect Bisulfite Kit (Qiagen) | Critical step requiring optimized conditions to minimize DNA degradation [4] |
| Methylation-Specific PCR Primers | Amplification of converted DNA with methylation discrimination | PyroMark PCR Kit (Qiagen), MSP primer design tools | Must be designed for bisulfite-converted sequence; avoid CpG sites in primer binding regions [4] |
| 5mC-Specific Antibodies | Immunoprecipitation or immunodetection of methylated DNA | Anti-5-methylcytosine (Abcam, Diagenode) | Used in MeDIP, m5C-RIP-seq; specificity validation essential [3] [7] |
| Methylated & Unmethylated Control DNA | Experimental controls for normalization | Millipore Sigma, Zymo Research | Critical for assay validation and quantitative accuracy [4] |
| Targeted Enrichment Systems | Capture of specific genomic regions | SureSelect Methyl-Seq (Agilent), SeqCap Epi (Roche) | Enables focused sequencing on regions of interest; reduces costs [3] |
| Long-read Sequencing Kits | Single-molecule sequencing with modification detection | Ligation Sequencing Kit (Oxford Nanopore) | Enables direct detection without bisulfite conversion; requires high molecular weight DNA [6] |
| Global Methylation Assay Kits | Quantification of total 5mC content | MethylFlash Global DNA Methylation ELISA Kit (EpiGentek) | Provides overall methylation levels but no locus-specific information [4] |
The comprehensive comparison of locus-specific versus genome-wide DNA methylation analysis methods reveals a complementary relationship rather than a competitive one between these approaches. Locus-specific methods like bisulfite pyrosequencing and amplicon bisulfite sequencing offer robust, cost-effective solutions for targeted biomarker validation and clinical diagnostics, demonstrating superior performance in standardized benchmarking studies [4]. Meanwhile, genome-wide approaches continue to provide powerful discovery platforms for identifying novel methylation patterns associated with biological processes and disease states [3].
Emerging technologies, particularly nanopore-based long-read sequencing, are beginning to bridge the gap between these approaches by enabling targeted yet comprehensive methylation analysis across large genomic regions with haplotype resolution [6]. This capability proves especially valuable for complex epigenetic phenomena such as genomic imprinting, where parent-of-origin methylation patterns regulate gene expression [6]. The ongoing development of more accessible, cost-effective, and multiplexed methylation analysis platforms will further accelerate the translation of epigenetic research into clinical applications, particularly in cancer diagnostics, neurological disorders, and developmental diseases.
As the field advances, the optimal selection of DNA methylation analysis methods will continue to depend on the specific research question, sample characteristics, and required balance between discovery power and quantitative precision. The experimental protocols and performance data presented here provide a framework for researchers to make informed decisions when investigating the biological roles of 5-methylcytosine in health and disease.
The analysis of DNA methylation, a key epigenetic modification, is fundamental to advancing our understanding of gene regulation, cellular differentiation, and disease mechanisms. Within this field, a fundamental methodological divide separates two distinct approaches: locus-specific analysis and genome-wide profiling. Each paradigm serves fundamentally different objectives, employs distinct technical frameworks, and answers unique biological questions. Locus-specific methods focus on deep interrogation of predefined genomic regions of interest, offering quantitative precision for clinical assay development and validation. In contrast, genome-wide approaches provide an unbiased exploration of epigenetic landscapes across entire genomes, enabling discovery-driven research without preexisting hypotheses about specific genomic locations. This methodological dichotomy reflects the complementary needs of targeted biomarker validation versus exploratory epigenomic mapping, both essential for progress in basic research and clinical applications. The choice between these approaches has profound implications for experimental design, resource allocation, and biological interpretation, making a clear understanding of their respective objectives, capabilities, and limitations essential for researchers, scientists, and drug development professionals working in the field of epigenetics [8] [9].
Core Objectives and Applications
Genome-Wide DNA Methylation Analysis
Genome-wide DNA methylation analysis aims to provide an unbiased, comprehensive survey of methylation patterns across the entire genome without prior assumptions about which regions might be biologically significant. This hypothesis-generating approach enables the discovery of novel differentially methylated regions (DMRs) associated with developmental processes, disease states, or environmental exposures. By covering extensive genomic territories, these methods can identify methylation markers and signatures that would be impossible to predict from existing knowledge [8] [10].
The primary applications of genome-wide analysis include epigenome-wide association studies (EWAS), which correlate methylation patterns with specific phenotypes or exposures across the entire genome; de novo biomarker discovery for various diseases, particularly cancer; and basic research into fundamental epigenetic mechanisms across different biological systems [11]. For example, large-scale comparative studies across 580 animal species utilized reduced representation bisulfite sequencing (RRBS) to establish evolutionary patterns of DNA methylation, demonstrating the power of genome-wide approaches for broad comparative biology [11]. Similarly, in oncology, genome-wide techniques have identified characteristic hypermethylation of CpG islands in cancer cells, leading to the discovery of novel tumor suppressor genes silenced by epigenetic mechanisms [12] [10].
Locus-Specific DNA Methylation Analysis
Locus-specific DNA methylation analysis focuses on quantifying methylation levels at predefined genomic regions with high precision and sensitivity. This hypothesis-driven approach validates candidate biomarkers identified through discovery-phase studies and translates them into clinically applicable assays. The core objective is to obtain accurate, reproducible methylation measurements at specific loci known to have biological or clinical significance [13] [9].
The primary applications of locus-specific analysis include clinical diagnostics, where defined methylation markers are used for disease detection, classification, or prognosis; biomarker validation following genome-wide discovery studies; and longitudinal monitoring of methylation patterns in response to therapy or disease progression [13] [14]. For instance, the EpiClass method was developed specifically for liquid biopsy applications, leveraging statistical differences in single-molecule methylation density distributions at the ZNF154 locus to detect ovarian carcinoma in circulating cell-free DNA [13]. In neurodevelopmental disorders, single-locus methylation tests at the NSD1 CpG island have been implemented for streamlined molecular diagnosis of Sotos syndrome, demonstrating the clinical utility of focused methylation analysis [14].
Technical Methodologies and Workflows
Genome-Wide Profiling Technologies
Genome-wide DNA methylation analysis employs several major technological platforms, each with distinct principles, advantages, and limitations. These methods can be broadly categorized into three groups: bisulfite conversion-based, enzyme digestion-based, and affinity enrichment-based approaches [8] [10].
Whole-genome bisulfite sequencing (WGBS) represents the gold standard for comprehensive methylation mapping, providing single-base resolution methylation measurements across virtually all CpG sites in the genome. The method works by treating DNA with bisulfite, which converts unmethylated cytosines to uracils (read as thymines in sequencing) while leaving methylated cytosines unchanged. After sequencing, methylation status is determined by comparing the sequence to an untreated reference [15] [10]. Although WGBS theoretically covers the entire genome, in practice it assesses approximately 50% of the genome, with biases toward regions with ≥2 CpGs/100 bp [15] [16]. The main limitations include high sequencing costs, substantial DNA input requirements, and complex bioinformatics challenges due to reduced sequence complexity after bisulfite conversion [15].
Reduced representation bisulfite sequencing (RRBS) provides a more targeted and cost-effective alternative by using restriction enzymes (typically MspI) to digest genomic DNA and select for CpG-rich regions, followed by bisulfite sequencing. This approach covers approximately 1-2 million CpG sites in the human genome, focusing on CpG islands, promoters, and other regulatory elements with high CpG density [15] [11]. RRBS is particularly valuable for large-scale comparative studies, as demonstrated by its application in profiling 580 animal species, where it effectively captured methylation patterns in conserved regulatory regions across diverse taxa [11]. A significant limitation is its bias toward high CpG density regions (≥3 CpGs/100 bp), covering only about 20% of the genome [15] [16].
Methylated DNA immunoprecipitation sequencing (MeDIP-seq) utilizes an antibody against 5-methylcytosine to immunoprecipitate methylated DNA fragments, followed by sequencing. This enrichment-based approach is particularly effective for regions with low to moderate CpG density (<5 CpGs/100 bp), which constitute >95% of the genome [15] [16] [10]. MeDIP-seq provides higher genome coverage than RRBS but offers lower resolution (100-300 bp) and cannot distinguish methylation at individual CpG sites. It is also unable to discriminate between different cytosine modifications (e.g., 5mC vs. 5hmC) [10].
Locus-Specific Analysis Technologies
Locus-specific DNA methylation analysis employs targeted methods that focus on predetermined genomic regions with clinical or biological significance. These techniques prioritize accuracy, sensitivity, and practical implementation for diagnostic applications [13] [9].
Bisulfite pyrosequencing combines bisulfite conversion with sequencing by synthesis to quantitatively determine methylation levels at individual CpG sites within a targeted amplicon. The method provides high accuracy (typically ±5-10%) and reproducibility, making it one of the most widely used techniques for validation of methylation biomarkers [9]. Its quantitative nature, relatively low cost, and compatibility with standard laboratory equipment have established it as a workhorse for targeted epigenetic analysis in both research and clinical settings.
Methylation-specific PCR (MSP) and its quantitative variant (qMSP) use primers designed to specifically amplify either methylated or unmethylated DNA sequences after bisulfite conversion. This method offers exceptional sensitivity for detecting rare methylated alleles in background of unmethylated DNA, making it particularly suitable for liquid biopsy applications where tumor-derived methylated DNA represents only a small fraction of total circulating DNA [13] [9]. A community-wide benchmarking study found that qMSP showed excellent performance for detecting methylated DNA fragments in an excess of unmethylated fragments, though it provides only semiquantitative measurements of methylation levels [9].
Digital methylation-specific analysis approaches, such as the DREAMing (Digital Methylation Analysis) method, provide single-molecule resolution by assessing the methylation status of individual DNA molecules. This digital approach enables precise quantification of methylation patterns and can detect mosaic methylation or heterogeneous cell populations [13]. The EpiClass algorithm leverages such digital methylation density distributions to improve cancer detection in liquid biopsies, demonstrating how advanced analysis of locus-specific data can enhance diagnostic performance [13].
Emerging technologies like nanopore sequencing enable direct detection of DNA methylation without bisulfite conversion by measuring electrical signal changes as DNA passes through protein nanopores. This approach allows simultaneous genetic and epigenetic analysis from a single sequencing run and can be adapted for both genome-wide and locus-specific applications through targeted enrichment strategies [14]. Recent studies have successfully used nanopore sequencing to identify disease-specific methylation signatures (episignatures) for neurodevelopmental disorders like Sotos syndrome, highlighting its potential for integrated genetic-epigenetic diagnostics [14].
Comparative Performance Analysis
Technical Specifications and Capabilities
The fundamental differences between genome-wide and locus-specific DNA methylation analysis methods are reflected in their technical specifications, genomic coverage, and resolution characteristics. Understanding these parameters is essential for selecting the appropriate methodological approach for specific research questions or clinical applications.
Table 1: Technical Comparison of Major DNA Methylation Analysis Methods
| Method | Resolution | Genomic Coverage | DNA Input | Primary Applications | Key Limitations |
|---|---|---|---|---|---|
| WGBS | Single-base | ~50% of genome [15] | 100-1000 ng [10] | Reference epigenomes, discovery studies | High cost, complex bioinformatics, sequence alignment issues [15] |
| RRBS | Single-base | ~20% of genome (high CpG density) [15] [16] | 10-100 ng [11] | Comparative studies, cancer biomarker discovery | Bias toward high CpG density regions, misses hypomethylated areas [15] |
| MeDIP-seq | 100-300 bp | >95% of genome (low CpG density) [15] [16] | 1-100 ng [10] | Tissue-specific methylation, global patterns | Cannot distinguish individual CpGs, antibody specificity issues [10] |
| Bisulfite Pyrosequencing | Single-base | Predefined loci | 10-100 ng [9] | Biomarker validation, clinical assays | Limited to targeted regions, requires primer design |
| qMSP | Pattern-based | Predefined loci | 1-10 ng [9] | Liquid biopsy, minimal residual disease detection | Semiquantitative, prone to amplification bias [9] |
| Nanopore Sequencing | Single-base | Flexible (targeted to whole genome) [14] | 100-1000 ng [14] | Integrated genetic-epigenetic analysis, diagnostics | Emerging technology, standardization ongoing [14] |
Quantitative Performance Metrics
Independent benchmarking studies have provided comprehensive comparisons of the quantitative performance of various DNA methylation analysis methods. A community-wide evaluation involving 18 laboratories assessed 27 different methylation assays for their sensitivity, reproducibility, and ability to discriminate between biological samples [9].
The study found that bisulfite pyrosequencing and amplicon bisulfite sequencing demonstrated the best all-round performance across multiple metrics, including accuracy, reproducibility, and robustness to sample quality variations. These methods showed high concordance with established gold standards and performed well across diverse genomic contexts, including regions with different CpG densities and GC content [9].
For detection of low-level methylation in mixed samples, as encountered in liquid biopsy applications, digital approaches and qMSP showed superior sensitivity. The EpiClass method, which analyzes methylation density distributions at single-molecule resolution, achieved 91.7% sensitivity and 100% specificity for detecting ovarian carcinoma in plasma samples, outperforming the standard CA-125 test [13].
Table 2: Performance Characteristics in Clinical Application Contexts
| Application Context | Recommended Method(s) | Achievable Performance | Considerations |
|---|---|---|---|
| Novel biomarker discovery | WGBS, RRBS, MeDIP-seq | Genome-wide coverage, identification of DMRs | Trade-off between coverage and cost; RRBS balances both well [15] [11] |
| Biomarker validation | Bisulfite pyrosequencing, AmpliconBS | High accuracy (±5-10%), excellent reproducibility [9] | Gold standard for quantitative validation |
| Liquid biopsy cancer detection | qMSP, Digital analysis (EpiClass) | Sensitivity >90%, specificity >90% for EOC detection [13] | Digital methods better for heterogeneous samples |
| Integrated genetic-epigenetic testing | Nanopore sequencing | Simultaneous variant and methylation detection [14] | Emerging technology with promising one-stop potential |
| Large-scale clinical screening | Pyrosequencing, MSP | High throughput, cost-effective for targeted loci [9] | Established protocols, widely available |
Experimental Protocols and Research Reagents
Genome-Wide Methylation Profiling with RRBS
The Reduced Representation Bisulfite Sequencing (RRBS) protocol provides a cost-effective approach for genome-wide methylation analysis that focuses on CpG-rich regions, making it particularly suitable for large-scale comparative studies [11].
Key Protocol Steps:
- DNA Digestion: Digest genomic DNA with MspI restriction enzyme (recognition site: CCGG), which cuts regardless of methylation status, generating fragments with defined ends [11].
- Size Selection: Perform size selection (typically 40-220 bp) to enrich for CpG-rich fragments using magnetic bead-based cleanups [15].
- End Repair and Adenylation: Repair fragment ends and add A-overhangs for adapter ligation.
- Adapter Ligation: Ligate methylated sequencing adapters to fragments.
- Bisulfite Conversion: Treat adapter-ligated DNA with bisulfite reagent using optimized conversion kits (e.g., EZ DNA Methylation kits) [11].
- PCR Amplification: Amplify converted DNA with polymerase capable of reading uracils.
- Library Validation and Sequencing: Validate library quality and sequence on appropriate platform (Illumina recommended).
Critical Considerations:
- Input DNA: 10-100 ng of high-quality genomic DNA
- Include unmethylated lambda phage DNA as conversion control
- Use duplicate processing to assess technical variability
- Bioinformatics processing requires specialized tools (Bismark, BS-Seeker2) for alignment and methylation calling [15]
Targeted Locus Analysis with EpiClass/DREAMing
The DREAMing (Digital Methylation Analysis) workflow combined with the EpiClass algorithm provides a highly sensitive approach for detecting cancer-specific methylation in liquid biopsies through quasi-digital analysis of methylation density [13].
Key Protocol Steps:
- Cell-free DNA Extraction: Isolate cfDNA from plasma samples (1-3 mL plasma typically yields sufficient DNA).
- Targeted Amplification: Amplify target locus (e.g., ZNF154) from bisulfite-converted DNA with primers flanking region of interest.
- High-Resolution Melting Analysis: Perform HRM under controlled conditions to distinguish epialleles based on melting temperature.
- Digital Analysis: Deconvolute complex melting profiles to determine methylation density of individual molecules.
- EpiClass Analysis: Apply classification algorithm to methylation density distributions to distinguish case from control samples.
Critical Parameters:
- Analytical threshold: Optimize based on expected ctDNA fraction (typically 0.1-1% in early cancer)
- Control samples: Include both positive (methylated) and negative (unmethylated) controls
- Sample classification: Determine optimal methylation density and fraction thresholds using training cohort [13]
Research Reagent Solutions
Table 3: Essential Research Reagents for DNA Methylation Analysis
| Reagent/Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Bisulfite Conversion Kits | EZ DNA Methylation kits, CpGenome Turbo Kit | Chemical conversion of unmethylated C to U | Conversion efficiency >99% required; assess with unmethylated controls [9] |
| Methylation-Specific Enzymes | MspI, TaqI, McrBC | Restriction digestion for RRBS or CHARM | Enzyme lot variability; optimize digestion conditions [15] [10] |
| Methylation Detection Antibodies | Anti-5-methylcytosine, MBD proteins | Enrichment of methylated DNA for MeDIP/MBDCap | Antibody specificity critical; validate with spike-in controls [10] |
| PCR Reagents | Bisulfite-converted DNA polymerases, HOT START varieties | Amplification of converted DNA | Uracil-tolerant polymerases essential for bisulfite-converted templates [9] |
| Methylation Standards | Fully methylated/unmethylated control DNA, synthetic oligos | Quantification controls, standard curves | Commercial sources available; verify methylation status independently |
| Library Preparation Kits | Illumina RRBS kits, Accel-NGS Methyl-Seq | Sequencing library construction | Method-specific compatibility; size selection critical for RRBS [15] |
The fundamental divide between locus-specific and genome-wide DNA methylation analysis reflects complementary rather than competing approaches in epigenetic research. Each addresses distinct objectives: genome-wide methods enable unbiased discovery of novel methylation patterns across the entire epigenetic landscape, while locus-specific techniques provide the precision and sensitivity required for clinical assay development and validation. The choice between these paradigms depends fundamentally on the research question, with discovery-phase studies benefiting from the comprehensive coverage of WGBS or RRBS, and translational applications requiring the quantitative accuracy of bisulfite pyrosequencing or digital methylation analysis [8] [13] [9].
Emerging technologies are beginning to bridge this methodological divide. Single-cell methylome analysis now enables genome-wide profiling at cellular resolution, revealing previously unappreciated heterogeneity in epigenetic regulation [8]. Long-read sequencing technologies, particularly nanopore and PacBio platforms, offer simultaneous genetic and epigenetic analysis from a single experiment, potentially unifying discovery and application in a single workflow [14]. These advances, coupled with improved computational methods for analyzing methylation heterogeneity and pattern conservation, promise to further enhance both our fundamental understanding of epigenetic regulation and our ability to translate this knowledge into clinically actionable biomarkers [13] [11].
For researchers and drug development professionals, the current landscape offers a diverse toolkit for DNA methylation analysis. The strategic selection of appropriate methods—whether genome-wide for discovery or locus-specific for validation—remains essential for advancing both basic science and clinical applications in the rapidly evolving field of epigenetics.
The analysis of DNA methylation, a key epigenetic modification, is crucial for understanding gene regulation, cellular differentiation, and the pathogenesis of various diseases, including cancer [17] [18]. The choice of technology fundamentally shapes the scope, resolution, and biological validity of epigenetic research and clinical diagnostics. This guide provides an objective comparison of the three principal technological principles underlying DNA methylation analysis: bisulfite conversion, enzymatic methods, and affinity enrichment. Each method operates on a distinct biochemical principle to discriminate methylated from unmethylated cytosine, with significant implications for data output, applicability to different research questions, and compatibility with sample types such as liquid biopsies [19]. Framed within the broader thesis of locus-specific versus genome-wide analysis, this review equips researchers and drug development professionals with the data needed to select the optimal technological path for their specific experimental and clinical objectives.
Core Technological Principles and Workflows
Bisulfite Conversion
Bisulfite conversion is widely regarded as the gold-standard method for detecting DNA methylation at single-base resolution [18] [20]. In this chemical process, genomic DNA is treated with sodium bisulfite, which selectively deaminates unmethylated cytosines into uracils. These uracils are then read as thymines during subsequent PCR amplification and sequencing. In contrast, methylated cytosines (5-methylcytosine, 5mC) are protected from this conversion and remain as cytosines [21]. The fundamental workflow involves denaturing the DNA, treating it with sodium bisulfite, and then desalting and purifying the converted DNA for downstream analysis [20]. A key limitation is that this method cannot distinguish between 5mC and 5-hydroxymethylcytosine (5hmC), as both resist conversion [21] [18]. Furthermore, the process is known to cause significant DNA fragmentation and loss, which poses a challenge for low-quality or low-quantity samples like cell-free DNA (cfDNA) [20].
Enzymatic Methods
Enzymatic conversion has emerged as a more recent alternative designed to overcome the drawbacks of bisulfite conversion [20]. This approach uses a series of enzymes to achieve a similar end result. First, a TET enzyme oxidizes 5mC, and a T4-BGT glycosyltransferase glucosylates 5hmC, effectively "protecting" these modified cytosines. Subsequently, an APOBEC enzyme deaminates the unmethylated cytosines into dihydrouracil (DHU), which is replaced by thymine during PCR [20]. The enzymatic process is gentler on DNA, resulting in significantly less fragmentation and making it more suitable for degraded samples like forensic evidence or cfDNA [20]. While the conversion principle is similar, the enzymatic nature of the steps differentiates its performance and optimal application scenarios.
Affinity Enrichment
Affinity enrichment methods take a fundamentally different approach by using proteins or antibodies to physically pull down methylated DNA fragments, rather than altering the DNA sequence itself. Two primary techniques are employed: Methylated DNA Immunoprecipitation (MeDIP), which uses an antibody against 5-methylcytosine to enrich for methylated DNA [18] [22]; and Methyl-CpG Binding Domain (MBD) enrichment, which uses recombinant proteins containing the MBD domain to capture DNA fragments with methylated CpGs [23] [18]. These methods are particularly effective for enriching highly methylated genomic regions, such as promoter CpG Islands when they are silenced. However, a major limitation is that they do not provide single-base resolution; instead, they indicate the relative methylation level of an entire captured fragment [18]. They are also cost-effective for genome-wide screening when the highest resolution is not required [22].
The workflows for these three core principles are illustrated below.
Comprehensive Technology Comparison Table
The following table summarizes the key characteristics, advantages, and limitations of the three core technologies, providing a direct comparison to guide method selection.
Table 1: Comparative overview of DNA methylation analysis technologies
| Feature | Bisulfite Conversion | Enzymatic Methods | Affinity Enrichment |
|---|---|---|---|
| Underlying Principle | Chemical deamination of unmethylated C [21] | Enzymatic protection and deamination [20] | Immunoprecipitation or protein binding [23] [18] |
| Resolution | Single-base [21] [18] | Single-base [20] | Regional (100-500 bp) [18] |
| DNA Input | 500 pg - 2 µg [20] | 10 - 200 ng [20] | Varies, often high (µg range) [24] |
| DNA Damage | High fragmentation and loss [20] | Low fragmentation [20] | Minimal (native DNA) |
| Distinguishes 5mC/5hmC | No [21] [18] | Yes (with modification) [18] | Possible with specific antibodies [22] |
| Primary Advantage | Gold standard, high resolution | Gentle on DNA, suitable for cfDNA | Cost-effective for genome-wide screening |
| Key Limitation | DNA degradation, sequence complexity reduction | Lower converted DNA recovery [20] | No single-base resolution |
| Best For | Targeted analysis, single-base resolution | Low-quality/quantity samples (e.g., liquid biopsies) [20] | Genome-wide methylation profiling |
Experimental Performance Data and Protocols
Quantitative Performance Benchmarking
Independent comparative studies provide critical performance metrics for objective technology selection. A 2025 developmental validation study directly compared a leading bisulfite conversion kit (Zymo Research EZ DNA Methylation) with the primary enzymatic conversion kit (NEBNext Enzymatic Methyl-seq Conversion Module) using a multiplex qPCR assay (qBiCo) to measure key parameters [20].
Table 2: Experimental performance data for conversion methods (10 ng DNA input)
| Performance Metric | Bisulfite Conversion | Enzymatic Conversion |
|---|---|---|
| Conversion Efficiency | High (Limit of reproducible conversion: 5 ng) [20] | High (Limit of reproducible conversion: 10 ng) [20] |
| Converted DNA Recovery | Overestimated (130% reported) [20] | Low (40% reported) [20] |
| DNA Fragmentation (Index) | High (14.4 ± 1.2) [20] | Low-Medium (3.3 ± 0.4) [20] |
| Robustness on Degraded DNA | Low (High fragmentation) [20] | High (Gentler process) [20] |
This data indicates a key trade-off: while bisulfite conversion shows higher DNA recovery in assays, this may be an overestimation, and it comes at the cost of severe DNA fragmentation. Enzymatic conversion, while currently yielding lower recovery, is far gentler and creates higher-quality material from fragile samples, a crucial advantage for liquid biopsy analysis [20].
Detailed Experimental Protocols
Protocol for Bisulfite Conversion using a Commercial Kit
- Input: Use 10 ng - 2 µg of genomic DNA. For low-input samples, consider carrier RNA.
- Conversion: Dilute DNA in a total volume of 20 µL with nuclease-free water. Add 130 µL of CT Conversion Reagent, mix, and incubate in a thermal cycler (98°C for 8 minutes, 54°C for 60 minutes, hold at 4°C). This step denatures the DNA and performs the chemical deamination [20].
- Purification: Load the sample onto a Zymo-Spin IC Column containing M-Binding Buffer. Centrifuge at full speed (>10,000 rpm) for 30 seconds. Discard flow-through.
- Desalting and Wash: Add 100 µL of M-Wash Buffer to the column and centrifuge. Add 200 µL of M-Desulphonation Buffer, let stand at room temperature for 15-20 minutes, then centrifuge.
- Final Wash and Elution: Wash the column twice with 200 µL of M-Wash Buffer. Elute the converted DNA in 20 µL of M-Elution Buffer [20].
Protocol for Enzymatic Conversion
- Input: Use 10 - 200 ng of genomic DNA. Do not fragment prior to conversion for sensitive, low-input samples [20].
- Oxidation and Glycosylation: Set up the oxidation reaction and incubate at 37°C for 1 hour. Then, set up the glycosylation reaction and incubate at 37°C for 1 hour. These steps protect 5mC and 5hmC.
- Enzymatic Cleanup: Use a bead-based cleanup after each enzymatic step (oxidation and glycosylation). This is a critical but potentially tedious manual process [20].
- Deamination: Set up the deamination reaction and incubate at 37°C for 2 hours. This step converts unmethylated cytosines.
- Final Cleanup and Elution: Perform a final bead-based cleanup to inactivate the enzymes and elute the converted DNA in 20 µL of elution buffer [20].
Protocol for Affinity Enrichment (MeDIP)
- DNA Preparation: Shear 100 ng - 1 µg of genomic DNA to a fragment size of 100-500 bp using sonication or enzymatic digestion.
- Immunoprecipitation: Dilute the sheared DNA in IP buffer. Add an anti-5-methylcytosine antibody. Incubate the mixture overnight at 4°C with rotation [18].
- Capture and Wash: Add protein A/G magnetic beads to the mixture and incubate to capture the antibody-DNA complexes. Wash the beads several times with IP buffer to remove non-specifically bound DNA.
- Elution and Purification: Elute the enriched methylated DNA from the beads using a elution buffer (e.g., containing proteinase K). Purify the eluted DNA using a standard DNA cleanup kit [18].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these technologies relies on a suite of specialized reagents and kits.
Table 3: Key research reagent solutions for DNA methylation analysis
| Reagent / Kit Name | Function | Technology Principle |
|---|---|---|
| Zymo Research EZ DNA Methylation Kit [20] | Commercial kit for robust bisulfite conversion | Bisulfite Conversion |
| NEBNext Enzymatic Methyl-seq Conversion Module [20] | Commercial kit for gentle, enzymatic conversion | Enzymatic Conversion |
| Methylated DNA Immunoprecipitation (MeDIP) Kit | Immunoprecipitation of methylated DNA using 5mC antibody | Affinity Enrichment |
| MethylMiner Methylated DNA Enrichment Kit | Enrichment of methylated DNA using MBD protein | Affinity Enrichment |
| Anti-5-hydroxymethylcytosine (5hmC) Antibody [22] | Specific immunoprecipitation of hydroxymethylated DNA | Affinity Enrichment |
| APOBEC Enzyme [20] | Key enzyme for deaminating unmethylated C in EC | Enzymatic Conversion |
| TET / T4-BGT Enzymes [20] | Key enzymes for protecting 5mC/5hmC in EC | Enzymatic Conversion |
| Methylation-Specific PCR (MSP) Primers | For locus-specific analysis after conversion | Bisulfite Conversion |
| Illumina Infinium MethylationEPIC BeadChip [20] | Microarray for genome-wide analysis of ~850k CpGs | Bisulfite Conversion |
The selection of an appropriate DNA methylation analysis technology is a foundational decision that directly impacts research outcomes and diagnostic efficacy. Bisulfite conversion remains the established gold standard for applications demanding single-base resolution, despite its challenges with DNA degradation. Enzymatic methods represent a transformative advance, offering a gentler alternative that is particularly suited for the analysis of precious, low-quality samples like liquid biopsies, even if recovery rates need further optimization. Affinity enrichment provides a cost-effective path for genome-wide mapping where single-base resolution is not required. The choice between them is not a matter of superiority, but of alignment with specific research goals, sample characteristics, and analytical requirements. As the field moves towards multi-omics and clinical liquid biopsy applications, enzymatic and other direct detection methods are poised to play an increasingly prominent role in the epigenomic toolkit [19].
Choosing the appropriate method for DNA methylation analysis is a critical step in epigenetic research, as the choice directly impacts the validity and scope of the biological conclusions. The decision is primarily guided by the experimental aim (genome-wide discovery versus targeted analysis), the nature of the available samples, and budgetary constraints. This guide provides a structured comparison of current technologies, supported by experimental data and workflows, to inform researchers and drug development professionals.
Table of Contents
- Technology Comparison at a Glance
- Experimental Protocols for Key Methods
- Decision Workflow for Method Selection
- Essential Research Reagent Solutions
Technology Comparison at a Glance
The following tables summarize the core characteristics of prevalent DNA methylation analysis methods, categorizing them by their primary application.
Table 1: Comparison of Discovery-Focused Genome-Wide Methods
| Method | Key Principle | Resolution | Best For | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Whole-Genome Bisulfite Sequencing (WGBS) [25] [26] | Bisulfite conversion & NGS | Single-base | Gold standard for base-resolution methylome; non-model organisms. | Single-nucleotide resolution; covers practically every cytosine. | High cost; computationally intensive; significant DNA loss from bisulfite treatment [25] [27]. |
| Reduced Representation Bisulfite Sequencing (RRBS) [11] | Methylation-sensitive restriction enzyme & bisulfite sequencing | Single-base | Cost-effective profiling of CpG-rich regions; large cohort studies. | Cost-effective; requires less sequencing than WGBS; enriches for regulatory CpG islands. | Bias towards CpG-rich regions (promoters, CpG islands); does not cover entire genome [11]. |
| Methylation Microarrays (e.g., Infinium) [28] [9] | Bisulfite conversion & array hybridization | Pre-defined sites | High-throughput analysis of large sample cohorts; EWAS. | High reproducibility; streamlined workflow; low cost per sample; validated for FFPE samples [28]. | Limited to pre-designed CpG sites (e.g., 3,000-850,000); not for novel discovery outside array content [28]. |
| Affinity Enrichment (MeDIP/MBD) [25] [29] | Antibody-based enrichment of methylated DNA | Regional | Low-cost genome-wide screening; labs skilled in ChIP-seq. | Lower cost than bisulfite sequencing; straightforward protocol. | Lower resolution; bias from copy number variation, GC content, and CpG density [25]. |
Table 2: Comparison of Targeted DNA Methylation Analysis Methods
| Method | Key Principle | Resolution | Best For | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Amplicon Bisulfite Sequencing (AmpliconBS) [9] | PCR of bisulfite-converted DNA & NGS | Single-base | Validating DMRs; analyzing specific loci (e.g., gene promoters). | High sensitivity and accuracy; cost-effective for a few targets. | Throughput limited by PCR reactions; primer design can be challenging. |
| Bisulfite Pyrosequencing (Pyroseq) [24] [9] | Sequencing-by-synthesis of bisulfite-converted PCR products | Single-base | Validating and quantifying methylation at a few CpGs; high reproducibility. | Excellent quantitative accuracy; high throughput; rapid turnaround. | Limited to short sequences (~100-200bp); not ideal for large genomic regions. |
| Targeted Methyl-Seq (Capture) [27] | Bisulfite conversion & hybrid capture | Single-base | Profiling methylation in specific gene panels (e.g., cancer biomarkers). | Focuses resources; high coverage on targets; cost-effective for large genomic regions. | Custom panel design required; probe design must account for bisulfite-converted sequence [27]. |
| Methylation-Specific PCR (qMSP) [9] | PCR with primers specific to methylated sequence | Locus-specific | Clinical biomarker detection; high-sensitivity detection of rare methylated alleles. | High sensitivity and specificity; fast and simple. | Semi-quantitative; requires careful optimization and controls. |
Table 3: Practical Considerations: Sample Input, Cost, and Skill Requirements
| Method | Recommended DNA Input | Sample Quality | Relative Cost | Bioinformatic Complexity |
|---|---|---|---|---|
| WGBS | Low (pg–ng scale) [25] | High quality recommended | Very High | Very High |
| RRBS | Not specified in results | High quality recommended | Medium | High |
| Methylation Array | Varies by platform | Robust (including FFPE) [28] | Low (per sample) | Low |
| Affinity Enrichment | Higher than bisulfite methods [25] | High quality recommended | Low | Medium |
| AmpliconBS / Pyroseq | Varies (50-200ng typical) | Robust (including FFPE) | Low | Low to Medium |
| Targeted Methyl-Seq | Varies (can be low input) | Robust (including FFPE and cfDNA) [27] | Medium (depends on target size) | Medium |
Experimental Protocols for Key Methods
Whole-Genome Bisulfite Sequencing (WGBS)
WGBS is the gold standard for unbiased, genome-wide methylation profiling at single-base resolution [25].
Detailed Protocol:
- DNA Fragmentation: Genomic DNA is mechanically sheared to a desired fragment size (e.g., 200-500bp).
- Library Preparation: Sequencing adapters are ligated to the fragmented DNA. This can be performed before or after bisulfite conversion.
- Bisulfite Conversion: DNA is treated with sodium bisulfite, which deaminates unmethylated cytosines to uracils, while methylated cytosines remain unchanged [25]. Converted uracils are read as thymines during sequencing.
- PCR Amplification & Sequencing: The converted DNA is amplified and subjected to next-generation sequencing on a platform such as Illumina [25] [26].
Key Consideration: Spiking-in an unmethylated control (e.g., λ-phage DNA) is critical to monitor conversion efficiency, which should routinely be >99% [25].
Amplicon Bisulfite Sequencing
This locus-specific method provides high-depth, quantitative methylation data for predefined regions.
Detailed Protocol:
- Bisulfite Conversion: Genomic DNA is converted with sodium bisulfite.
- PCR Primer Design: Primers are designed to anneal to the bisulfite-converted sequence, avoiding CpG sites to ensure unbiased amplification of methylated and unmethylated alleles.
- Target Amplification: The region of interest is PCR-amplified from the converted DNA.
- NGS Library Prep & Sequencing: Amplicons are prepared for sequencing, often by barcoding and pooling, then sequenced on an NGS platform [9].
Emerging Protocol: Enzymatic Methyl-Seq
An alternative to chemical bisulfite conversion that minimizes DNA damage.
Detailed Protocol:
- Enzymatic Conversion: DNA is treated with TET2 and APOBEC enzymes. TET2 oxidizes 5-methylcytosine, and APOBEC deaminates unmethylated cytosines to uracils [27].
- Library Prep & Sequencing: The converted DNA is then processed similarly to bisulfite-converted DNA for library preparation and sequencing. This method offers the advantage of minimal DNA loss due to less aggressive reaction conditions [27].
Decision Workflow for Method Selection
The following diagram outlines a logical pathway for selecting the most appropriate DNA methylation analysis method based on key project criteria.
Essential Research Reagent Solutions
Successful DNA methylation analysis relies on specialized reagents and kits. The following table details key solutions for major methodological steps.
Table 4: Key Research Reagent Solutions for DNA Methylation Analysis
| Reagent / Kit | Function | Application Notes |
|---|---|---|
| Sodium Bisulfite Conversion Kits (e.g., from Zymo Research) | Chemically converts unmethylated C to U; cornerstone of bisulfite-based methods. | Critical for high conversion efficiency (>99%); quality impacts all downstream data [25]. |
| Enzymatic Conversion Kits (e.g., TET2/APOBEC) | Enzymatically converts unmethylated C to U; alternative to bisulfite. | Minimizes DNA degradation; ideal for low-input or sensitive samples [27]. |
| Infinium MethylationEPIC v2.0 Kit (Illumina) | Microarray-based analysis of >900,000 CpG sites in the human genome. | For high-throughput epigenome-wide association studies (EWAS); reproducible and FFPE-compatible [28]. |
| Methylated DNA Immunoprecipitation (MeDIP) Kits | Uses anti-5mC antibody to pull down methylated DNA fragments for enrichment-based sequencing. | Lower-cost genome-wide method; requires higher DNA input than bisulfite methods [25] [29]. |
| Global DNA Methylation ELISA Kits (e.g., from Epigentek) | Immunoquantification of total 5mC content in a DNA sample. | Provides a rough, global estimate of methylation; high inter-assay variability [24]. |
| LINE-1 Pyrosequencing Assay | Amplifies and sequences bisulfite-converted LINE-1 retrotransposons. | Serves as a surrogate for estimating global DNA methylation hypomethylation [24]. |
| Targeted Methyl-Seq Panels (e.g., from Celemics) | Hybrid capture probes designed for bisulfite-converted sequences to enrich specific genomic regions. | Probes must account for C-to-T changes to ensure capturing efficiency; for focused studies [27]. |
| 5hmC Analysis Kits (oxBS-Seq, TAB-Seq) | Chemistries to distinguish 5-hydroxymethylcytosine (5hmC) from 5mC. | For advanced epigenetic profiling; requires specialized protocols and analysis [8] [26]. |
A Deep Dive into Techniques: From Targeted Assays to Genome-Scale Profiling
In the burgeoning field of epigenetics, DNA methylation analysis has bifurcated into two complementary approaches: genome-wide discovery and locus-specific validation. While genome-wide methods like whole-genome bisulfite sequencing (WGBS) and EPIC arrays excel at identifying novel methylation patterns across the entire genome, locus-specific techniques provide the quantitative precision, cost-effectiveness, and practical workflow required for biomarker validation and clinical application [30] [9]. This comparison focuses on three fundamental locus-specific workhorses—bisulfite pyrosequencing, methylation-sensitive high-resolution melting (MS-HRM), and quantitative methylation-specific PCR (qMSP)—which have proven indispensable for confirming epigenetic discoveries in large cohorts and translating them into clinically useful biomarkers.
The selection of an appropriate validation method represents a critical juncture in the research pipeline, balancing technical considerations like resolution and accuracy with practical concerns including throughput, cost, and implementation barrier. As large-scale epigenome-wide association studies continue to identify potential methylation biomarkers for various diseases, the role of these locus-specific methods becomes increasingly important [9]. Each technique offers distinct advantages and limitations, making them suited to different phases of the research and clinical development pathway. This guide provides an objective, data-driven comparison to inform researchers, scientists, and drug development professionals in selecting the optimal method for their specific application.
The three techniques represent different approaches to measuring DNA methylation at specific genomic loci after bisulfite conversion, which deaminates unmethylated cytosines to uracils while leaving methylated cytosines intact [31]. This fundamental treatment creates methylation-dependent sequence variations that form the basis for detection and quantification.
Table 1: Core Characteristics of Locus-Specific DNA Methylation Analysis Methods
| Characteristic | Bisulfite Pyrosequencing | MS-HRM | qMSP |
|---|---|---|---|
| Principle | Sequencing-by-synthesis with real-time light detection | Post-PCR analysis of melting curve behavior | Methylation-specific primer binding and amplification |
| Resolution | Single CpG dinucleotide | Regional (entire amplicon) | Regional (primer binding sites) |
| Quantification Type | Absolute quantification | Semi-quantitative to quantitative with standards | Relative quantification |
| Throughput | Medium to high | High | Very high |
| Cost per Sample | Medium | Low | Low to medium |
| DNA Quality Requirements | Standard to high | Flexible, works with degraded DNA | Flexible, works with low input DNA |
| Ease of Implementation | Medium (requires specialized instrument) | High (standard real-time PCR instrument) | High (standard real-time PCR instrument) |
Table 2: Performance Metrics Based on Experimental Comparisons
| Performance Metric | Bisulfite Pyrosequencing | MS-HRM | qMSP |
|---|---|---|---|
| Accuracy | High (considered gold standard) [32] | High (when calibrated) [33] | Medium (tendency for bias) [32] |
| Precision | Excellent (high reproducibility) [9] | Good [33] | Variable (primer-dependent) [31] |
| Sensitivity | Can detect 5% methylation [9] | Can detect 12.5% methylation [33] | High for detecting low methylation [32] |
| Dynamic Range | 0-100% [32] | 0-100% (with standards) [33] | Limited linear range [32] |
| Multiplexing Capability | Single amplicon per reaction | Single amplicon per reaction | Potential for multiple targets |
| Inter-assay Variability | Low (CV < 5%) [9] | Medium (CV 5-10%) [33] | High (CV can exceed 15%) [31] |
Bisulfite pyrosequencing provides quantitative methylation measurements at single-CpG resolution through sequential nucleotide dispensation and enzymatic light detection [31]. This technique excels when precise quantification of individual CpG sites is required, such as when different CpGs within the same region may have distinct biological implications. MS-HRM analyzes the melting behavior of PCR amplicons after bisulfite conversion, where methylated sequences retain more cytosines and thus exhibit higher melting temperatures [34]. This method offers the advantage of being a closed-tube system that minimizes contamination risk. qMSP utilizes primers specifically designed to amplify either methylated or unmethylated sequences after bisulfite conversion, providing a relative quantification approach that is highly sensitive but more prone to amplification bias [32].
Experimental Protocols and Workflows
Bisulfite Pyrosequencing Methodology
Bisulfite pyrosequencing employs a multi-step process beginning with sodium bisulfite conversion of DNA, which deaminates unmethylated cytosines to uracils while leaving methylated cytosines unchanged [31]. The converted DNA is then amplified using PCR with one biotinylated primer to enable subsequent immobilization. The resulting amplicon is bound to streptavidin-coated beads and converted to single-stranded DNA for sequencing. The core sequencing reaction involves sequential dispensation of nucleotides in a predefined order. When a nucleotide complements the template strand, DNA polymerase incorporates it, releasing pyrophosphate that converts to ATP via ATP sulfurylase, ultimately driving luciferase to produce measurable light [31]. Methylation percentage is quantitatively determined from the ratio of C (methylated) to T (unmethylated) signals at each CpG dinucleotide.
Bisulfite Pyrosequencing Workflow
Critical to this method is careful primer design to avoid CpG sites within primer binding regions, which could introduce amplification bias. Primers should be 20-30 base pairs long with melting temperatures around 60°C and contain at least four non-CpG cytosines to ensure specific amplification of bisulfite-converted DNA [31]. The sequencing read length typically ranges from 80-120 nucleotides, limiting analysis to relatively short regions, though multiple sequencing primers can be used for longer amplicons.
MS-HRM Protocol
MS-HRM begins with standard bisulfite conversion of DNA, followed by PCR amplification in the presence of a saturating DNA dye such as LCGreen or SYTO9 [33]. Following amplification, the product is gradually denatured by increasing temperature while continuously monitoring fluorescence. The resulting melting curves are analyzed by comparing sample curves to those from standards with known methylation ratios (typically 0%, 12.5%, 25%, 50%, 75%, and 100% methylated DNA) [33]. The temperature at which half of the DNA is denatured (melting temperature) and the shape of the melting curve provide information about the methylation status of the amplified region.
MS-HRM Workflow
Primer design for MS-HRM requires special consideration to create methylation-independent primers (MIP) that amplify both methylated and unmethylated sequences with similar efficiency [33]. This is achieved by placing non-CpG cytosines at the 3' end of primers to ensure they only bind to appropriately converted DNA. The amplicon size is typically kept small (80-150 bp) to ensure distinct melting profiles and to facilitate analysis of degraded DNA samples commonly encountered in clinical settings.
qMSP Methodology
qMSP utilizes methylation-specific primers that are designed to complement either the methylated sequence (after bisulfite conversion) or the unmethylated sequence [32]. The process begins with bisulfite conversion followed by real-time PCR amplification with fluorescence detection. The relative quantification of methylated DNA is typically normalized to a reference gene that is unmethylated or to total DNA input. The comparative Ct (ΔΔCt) method is commonly employed, where the difference in threshold cycles between the methylated target and reference is calculated relative to a calibrator sample [32].
qMSP Workflow
Primer design represents the most critical aspect of qMSP, as primers must specifically discriminate between methylated and unmethylated sequences after bisulfite conversion [31]. This is typically achieved by placing the 3' end of at least one primer at a CpG dinucleotide position. While this design provides high specificity, it can also introduce amplification bias if not carefully optimized. The inclusion of a unmethylated assay and reference genes is essential for accurate normalization and to account for variations in bisulfite conversion efficiency and DNA input [32].
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents for DNA Methylation Analysis
| Reagent/Kit | Function | Method Applicability | Key Considerations |
|---|---|---|---|
| Bisulfite Conversion Kits (e.g., EZ DNA Methylation Kit) | Converts unmethylated C to U while preserving 5mC | All three methods | Conversion efficiency critical; modern kits reduce DNA fragmentation [31] |
| Methylation Standards (0%, 50%, 100% methylated DNA) | Calibration and quality control | All three methods (essential for MS-HRM quantification) | Commercial standards available (e.g., EpiTect control DNA) [33] |
| Biotinylated Primers | Enables immobilization for pyrosequencing | Bisulfite Pyrosequencing | HPLC purification required to remove free biotin [31] |
| Saturation DNA Dyes (e.g., LCGreen, SYTO9) | Fluorescent detection of DNA melting | MS-HRM | Must not inhibit PCR; stable at high temperatures [33] |
| Pyrosequencing Kit and Cartridges | Sequencing reagents and reaction chambers | Bisulfite Pyrosequencing | Enzyme mixture, substrate, and nucleotides specifically formulated [31] |
| Methylation-Specific Primers | Selective amplification of methylated sequences | qMSP | Critical design parameter; 3' end at CpG sites enhances specificity [32] |
The choice between bisulfite pyrosequencing, MS-HRM, and qMSP depends heavily on research objectives, resource constraints, and intended applications. Pyrosequencing excels when precise quantification of individual CpG sites is required, as in diagnostic assay development or when analyzing regions with heterogeneous methylation patterns [9]. Its superior accuracy comes at the cost of specialized equipment and higher per-sample expense. MS-HRM represents an ideal compromise for many research settings, offering reliable semi-quantitative data with standard real-time PCR instrumentation at lower cost [33]. qMSP provides maximum sensitivity for detecting rare methylated alleles in background of unmethylated DNA, making it suitable for liquid biopsy applications, though it suffers from greater quantitative bias and more challenging assay optimization [32].
For clinical biomarker development, the field has observed a trend toward methods providing absolute quantification and single-CpG resolution. A community-wide benchmarking study evaluating 27 different assays found that bisulfite pyrosequencing and amplicon bisulfite sequencing demonstrated the best all-round performance for clinical applications [9]. However, for large-scale screening studies where cost-effectiveness is paramount, MS-HRM and qMSP offer practical advantages. Ultimately, these locus-specific methods will continue to play a critical role in translating epigenetic discoveries from basic research to clinical utility, with selection guided by the specific requirements of each application stage.
DNA methylation is a fundamental epigenetic mechanism involved in the regulation of gene expression, cellular differentiation, genomic imprinting, and the progression of complex diseases such as cancer and autoimmune disorders [8] [35]. For researchers and drug development professionals, selecting the appropriate method for genome-wide methylation analysis is crucial for study design, data quality, and interpretation. Two established technologies for this purpose are Whole-Genome Bisulfite Sequencing (WGBS) and the Illumina MethylationEPIC BeadChip microarray. WGBS is recognized for its comprehensive, base-resolution coverage of the methylome, whereas the EPIC array provides a cost-effective, targeted profiling solution [36] [37]. This guide objectively compares the performance, experimental protocols, and applications of these two standards, providing a structured framework to inform methodological selection in epigenetic research.
Whole-Genome Bisulfite Sequencing (WGBS) and the Illumina MethylationEPIC (EPIC) microarray represent two distinct philosophies in methylation profiling. WGBS employs next-generation sequencing (NGS) of bisulfite-converted DNA to assess methylation status at nearly every cytosine in the genome, offering unparalleled coverage [37] [38]. In contrast, the EPIC array uses hybridization to pre-designed probes to interrogate the methylation status of a pre-selected set of over 935,000 CpG sites, prioritizing cost-efficiency and ease of data analysis for large-scale studies [35].
The table below summarizes the core specifications and performance characteristics of these two methods.
Table 1: Key Specifications of WGBS and EPIC Microarray
| Feature | Whole-Genome Bisulfite Sequencing (WGBS) | Illumina MethylationEPIC Microarray |
|---|---|---|
| Fundamental Principle | Bisulfite conversion + NGS [39] | Hybridization to probe beads [37] |
| Resolution | Single-base [37] [38] | Single-CpG (for probed sites) [35] |
| Genomic Coverage | ~95% of CpGs in the human genome (~28 million sites) [38] | >935,000 pre-selected CpG sites (EPICv2) [35] |
| Typical DNA Input | ~1 µg (standard protocols); can be lower with specialized kits [37] [38] | 500 ng [37] |
| Key Strengths | Unbiased genome-wide coverage; detects methylation in non-CpG contexts, repetitive regions, and novel areas [37] [38] | Cost-effective for large cohorts; standardized, user-friendly workflow; high reproducibility [36] [35] |
| Key Limitations | High cost; complex data analysis; DNA degradation from bisulfite treatment [36] [37] [39] | Targeted, biased coverage; cannot discover novel methylation sites outside the panel [38] [35] |
| Best Suited For | Discovery-phase research, building reference methylomes, investigating uncharacterized genomic regions [38] | Large-scale epigenome-wide association studies (EWAS), clinical biomarker screening, population studies [36] [38] |
A comparative study evaluating WGBS, EPIC, and other emerging methods highlighted their complementary nature. Despite a substantial overlap in the CpG sites they detect, each method also captures unique sites, underscoring the importance of selecting a tool that aligns with the biological question [36]. For the EPIC array, this bias is intentional, as its probes are designed to cover regions of known regulatory function, such as promoters and enhancers [37] [35].
Performance and Experimental Data
Independent comparative studies have systematically assessed the performance of WGBS and EPIC arrays against each other and against benchmark technologies like whole-genome bisulfite sequencing.
Concordance and Coverage
The 2025 comparative evaluation by Epigenetics & Chromatin found that despite their technological differences, WGBS and EPIC arrays show strong agreement in the CpG sites they mutually cover [36]. The study reported that the enzymatic methyl-sequencing (EM-seq) method, which is chemically similar to WGBS, demonstrated the highest concordance with WGBS itself. The EPIC array, while covering a smaller fraction of the genome, provides highly reliable data for its targeted sites, making it a robust tool for focused hypotheses [36].
A critical consideration for WGBS is sequencing depth. A 2019 study in Scientific Reports provided a data-driven recommendation, indicating that a minimum of 30x coverage is required for reference methylomes, as per the International Human Epigenome Consortium (IHEC) standard. However, to achieve a level of precision broadly comparable to the methylation array, a minimum coverage of 100x is recommended [38].
Technical Reproducibility and DNA Input
The EPIC array exhibits high technical reproducibility, including for samples with DNA input levels below the manufacturer's recommendations [35]. Its standardized protocol and automated data processing pipelines contribute to its low technical variability, which is essential for detecting subtle methylation differences in large studies.
WGBS performance, on the other hand, can be influenced by the choice of library preparation kit. The same 2019 study compared three low-input kits—Swift, TruSeq, and QIAseq—on Illumina sequencing platforms. The Swift kit achieved the highest proportion of CpG sites assayed and effective coverage, while TruSeq suffered from the highest rate of PCR duplicates. The study also found that with updated Illumina software, a PhiX spike-in of only 5% is sufficient to generate high-quality WGBS data, reducing sequencing costs [38].
Table 2: Empirical Performance Metrics from Comparative Studies
| Performance Metric | WGBS | EPIC Microarray | Context and Notes |
|---|---|---|---|
| Coverage Precision | Lower precision at common depths (e.g., 30x); improves with higher coverage [38] | High precision for its targeted sites [38] | To match EPIC's precision, WGBS needs ~100x coverage [38] |
| Recommended Depth/Coverage | 100x for high precision [38] | >935,000 CpG sites (EPICv2) [35] | IHEC minimum standard is 30x for WGBS [38] |
| Library Prep Kits | Performance varies: Swift > TruSeq > QIAseq for CpG detection and low duplicate rates [38] | Not applicable (single standardized array) | Swift kit had highest insert size and lowest adapter contamination [38] |
| Input DNA Flexibility | Possible with low-input kits (e.g., ~20 ng for T-WGBS) [39] | Robust performance even with sub-optimal input [35] | T-WGBS: Tagmentation-based WGBS [39] |
Detailed Experimental Protocols
To ensure experimental reproducibility, researchers must adhere to standardized protocols. Below are the core methodologies for WGBS and EPIC array analyses as cited in the literature.
Whole-Genome Bisulfite Sequencing (WGBS) Protocol
The standard WGBS workflow involves multiple steps to convert, sequence, and analyze methylation patterns [37] [38] [39].
- DNA Extraction and Quality Control: High-molecular-weight DNA is extracted using kits such as the Nanobind Tissue Big DNA Kit or the DNeasy Blood & Tissue Kit. DNA purity is assessed via NanoDrop (260/280 and 260/230 ratios), and quantity is measured using a fluorometer (e.g., Qubit) [37].
- Library Preparation: Approximately 1 µg of DNA is fragmented, followed by end-repair and adenylation. Bisulfite conversion is performed using a kit like the EZ DNA Methylation Kit (Zymo Research). This critical step deaminates unmethylated cytosines to uracils, while methylated cytosines remain unchanged [37] [39]. After conversion, sequencing adapters are ligated, and the library is PCR-amplified. As noted in the performance section, the choice of library prep kit (e.g., Swift, TruSeq) significantly impacts outcomes [38].
- Sequencing: Libraries are sequenced on Illumina platforms (e.g., NovaSeq or HiSeq X) to a recommended depth of 100x for precision comparable to microarrays [38]. A low PhiX spike-in (e.g., 5%) is used to compensate for the reduced sequence complexity after bisulfite conversion [38].
- Bioinformatic Analysis: Raw sequencing reads are quality-trimmed using tools like Trim Galore! or Cutadapt. Processed reads are then aligned to a bisulfite-converted reference genome using aligners such as Bismark or Bowtie 2. Methylation calls are extracted by comparing cytosines in the sequenced reads to the reference, calculating the ratio of methylated to total reads for each cytosine [40] [38].
Illumina MethylationEPIC Array Protocol
The EPIC array workflow is more streamlined, leveraging the standardized Infinium chemistry [37] [35].
- DNA Extraction and Quality Control: Similar to WGBS, DNA is extracted and quantified. The standard input is 500 ng [37].
- Bisulfite Conversion and Amplification: DNA is bisulfite-converted using the EZ DNA Methylation Kit. The converted DNA is then whole-genome amplified, which enriches the fragments containing the CpG sites of interest [37].
- Hybridization, Staining, and Scanning: The amplified DNA is fragmented, precipitated, and resuspended before being applied to the EPIC BeadChip. The DNA hybridizes to locus-specific probes on the array. Subsequent single-base extension with fluorescently labeled nucleotides incorporates a dye that corresponds to the methylation status (methylated or unmethylated) of the CpG site. The BeadChip is then imaged with a scanner [37].
- Data Processing and Normalization: The image is processed using Illumina's software to generate intensity data (IDAT files). These files are imported into R/Bioconductor environments using packages like
minfifor quality control, normalization (e.g., using the beta-mixture quantile normalization method), and calculation of beta-values. Beta-values, ranging from 0 (unmethylated) to 1 (fully methylated), are the standard metric for methylation levels [37].
The following diagram illustrates the core workflows for both technologies, highlighting their parallel steps and key differences.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of DNA methylation profiling requires specific reagents and tools. The following table lists key solutions used in the protocols cited in this guide.
Table 3: Essential Research Reagents and Materials
| Item | Function | Example Products / Kits |
|---|---|---|
| DNA Extraction Kits | Isolate high-quality, high-molecular-weight DNA from various sample types. | Nanobind Tissue Big DNA Kit [37], DNeasy Blood & Tissue Kit (Qiagen) [37] |
| DNA Quantification Tools | Accurately measure DNA concentration and assess purity. | NanoDrop (Thermo Scientific) for purity, Qubit Fluorometer (Invitrogen) for quantity [37] |
| Bisulfite Conversion Kits | Chemically convert unmethylated cytosine to uracil for downstream detection. | EZ DNA Methylation Kit (Zymo Research) [37] |
| WGBS Library Prep Kits | Prepare NGS libraries from bisulfite-converted DNA; performance varies. | Swift Accel-NGS Methyl-Seq, Illumina TruSeq DNA Methylation Kit [38] |
| Methylation Microarray | The platform for hybridization-based methylation profiling. | Infinium MethylationEPIC v2.0 BeadChip (Illumina) [35] |
| NGS Sequencing Platforms | Generate sequence reads for WGBS libraries. | Illumina NovaSeq, Illumina HiSeq X [38] |
| Bioinformatic Tools | Process raw data, perform alignment, and extract methylation metrics. | WGBS: Bismark [40], Trim Galore!EPIC Array: minfi R package [37], ChAMP R package [37] |
WGBS and the Illumina EPIC array are both powerful technologies for genome-wide DNA methylation analysis, yet they serve different research needs. WGBS is the undisputed gold standard for comprehensiveness, offering base-resolution insight into nearly all CpGs in the genome, making it ideal for discovery-oriented research and the investigation of regions beyond predefined arrays [38]. This power, however, comes with higher costs, computational demands, and more complex protocols [36].
The EPIC array is a highly optimized tool for targeted profiling. Its strength lies in its cost-effectiveness, reproducibility, and streamlined workflow, which are essential for large-scale cohort studies and EWAS where the goal is to screen known regulatory regions across thousands of samples [36] [35]. Its main limitation is its inherent bias, as it cannot interrogate methylation patterns outside its designed probe set.
The choice between WGBS and EPIC is not a question of which is superior in absolute terms, but which is most appropriate for the specific research context. Scientists must weigh the requirements for discovery and resolution against the constraints of budget, sample size, and bioinformatic resources. As the field advances, emerging methods like enzymatic conversion sequencing (EM-seq) and long-read sequencing from Oxford Nanopore and PacBio [36] [40] are providing new alternatives that may overcome the limitations of both WGBS and microarrays, further expanding the epigenomic toolkit.
Enzymatic Methyl-Seq (EM-seq) and Long-Read Sequencing (ONT, PacBio)
DNA methylation is a fundamental epigenetic mechanism involved in gene regulation, cellular differentiation, and disease pathogenesis. For decades, bisulfite sequencing has been the gold standard for genome-wide methylation analysis, but its limitations—particularly DNA degradation and bias—have driven the development of alternative technologies. Enzymatic Methyl-Seq (EM-seq) and long-read sequencing platforms from Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) represent significant advancements that address these limitations while offering unique capabilities for locus-specific and genome-wide DNA methylation analysis. EM-seq employs an enzymatic conversion method that preserves DNA integrity, while long-read technologies enable direct detection of methylation patterns across extensive genomic regions. This guide provides an objective comparison of these emerging technologies, their performance metrics relative to established methods, and their applications in epigenetic research and drug development.
Enzymatic Methyl-Seq (EM-seq): Principle and Workflow
EM-seq utilizes a purely enzymatic approach to identify 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) at single-base resolution without the DNA damage associated with bisulfite treatment. The method employs three key enzymes in a two-step reaction process:
- Step 1 - Oxidation and Protection: TET2 enzyme oxidizes 5mC to 5-carboxylcytosine (5caC) through intermediate forms (5hmC and 5fC). Simultaneously, T4-BGT glucosylates genomic 5hmC to 5gmC, protecting both 5mC derivatives and 5hmC from subsequent deamination [41].
- Step 2 - Deamination: APOBEC3A deaminates unmodified cytosines to uracils, while all previously modified and protected cytosines remain unchanged [41].
- Sequencing and Analysis: During sequencing and alignment, unmethylated positions appear as thymines (T) while methylated positions are retained as cytosines (C), enabling methylation status determination [42].
The entire EM-seq library preparation process typically requires 2-4 days and is compatible with Illumina sequencing platforms [42].
Long-Read Sequencing Technologies
Oxford Nanopore Technologies (ONT)
ONT sequencing detects DNA methylation directly from native DNA without pre-treatment. The technology employs protein nanopores embedded in a synthetic membrane. As DNA passes through these pores, changes in electrical current are measured for each base. Modified bases, including 5mC and 5hmC, produce characteristic current deviations that distinguish them from unmodified bases [43]. Computational tools such as Nanopolish use these signal differences to call methylation status at CpG sites. ONT sequencing requires relatively high molecular weight DNA (approximately 1μg of 8kb fragments) but does not involve PCR amplification, preserving native modification patterns [44] [43].
Pacific Biosciences (PacBio)
PacBio's Circular Consensus Sequencing (CCS) detects DNA methylation through kinetic analysis during the sequencing process. The technology uses hairpin adapters to create single-stranded circular templates that are sequenced repeatedly. Modified bases alter polymerase kinetics, measurable as changes in Interpulse Duration (IPD) and Pulse Width (PW) [45]. Tools like ccsmeth employ deep learning models (bidirectional GRU with attention networks) to process these kinetic features and predict methylation states at single-molecule resolution [45]. PacBio sequencing is particularly valuable for methylation phasing and detecting allele-specific methylation across long genomic stretches.
Performance Comparison and Experimental Data
Comprehensive Technology Comparison
Table 1: Comparative Analysis of DNA Methylation Detection Technologies
| Parameter | EM-seq | WGBS | ONT | PacBio | EPIC Array |
|---|---|---|---|---|---|
| Resolution | Single-base | Single-base | Single-base | Single-base | Pre-defined sites only |
| DNA Input | 100 pg - 10 ng [41] | 100 ng+ [42] | ~1 μg [44] | Varies | 500 ng [44] |
| Coverage Uniformity | Even GC distribution [41] | GC bias [42] | No GC bias [42] | No GC bias | Probe-dependent |
| DNA Degradation | Minimal [42] | Significant [42] [44] | None | None | Moderate |
| Read Length | Short-read (Illumina) | Short-read | Long-read (kb+) | Long-read (≥10 kb) [45] | N/A |
| 5hmC Detection | Yes [41] | No (without oxBS) [43] | Yes [43] | Yes [45] | Indirectly |
| CpG Sites Covered | ~28 million (human) | ~28 million (human) | ~27 million (human) [43] | ~28 million (human) | ~935,000 [42] |
| Cost | Higher than WGBS [42] | Low | High | High | Low |
Performance Metrics from Experimental Studies
EM-seq vs. WGBS and PBAT
In a systematic comparison using Arabidopsis thaliana, EM-seq demonstrated significantly higher detection sensitivity than WGBS for low-input DNA (10ng), detecting 32% more methylation sites across CG, CHG, and CHH contexts [42]. The misjudgment rate of methylation status with EM-seq was only 2.1% under low-input conditions, nearly 64% lower than WGBS's 5.8% [42].
When compared to Post-Bisulfite Adapter Tagging (PBAT) for low-input DNA (10ng), EM-seq showed a 25% higher library conversion rate and identified 18% more rare methylation sites at CHG and CHH contexts [42]. Both methods showed high reproducibility with intra-group correlation coefficients (ICC) >0.85 [42].
Long-Read Sequencing Performance
For ONT sequencing, a comprehensive analysis of 7,179 human blood samples demonstrated high accuracy in CpG methylation detection when compared to oxidative bisulfite sequencing (oxBS). The Pearson correlation coefficient for average 5-mCpG rates was 0.9594 between ONT and oxBS, with a mean absolute difference of 0.0471 per CpG [43]. Coverage significantly impacted consistency, with sequencing at ≥20× coverage yielding optimal results [43].
For PacBio CCS, the ccsmeth tool demonstrated high accuracy in 5mCpG detection, achieving 0.90 accuracy and 0.97 Area Under the Curve at single-molecule resolution with long (≥10kb) CCS reads [45]. At the genome-wide site level, ccsmeth achieved >0.90 correlations with bisulfite sequencing using only 10× reads [45].
Table 2: Quantitative Performance Metrics from Comparative Studies
| Comparison | Metric | Result | Context |
|---|---|---|---|
| EM-seq vs. WGBS [42] | Sites detected | 32% more with EM-seq | Low-input DNA (10ng) |
| EM-seq vs. WGBS [42] | Misjudgment rate | 2.1% (EM-seq) vs 5.8% (WGBS) | Low-input conditions |
| EM-seq vs. PBAT [42] | Library conversion | 25% higher with EM-seq | 10ng DNA input |
| EM-seq vs. PBAT [42] | Rare CHG/CHH sites | 18% more with EM-seq | Methylation context |
| ONT vs. oxBS [43] | Pearson correlation | 0.9594 | CpG average |
| ONT vs. oxBS [43] | Mean absolute difference | 0.0471 | Per CpG |
| PacBio ccsmeth [45] | Read-level accuracy | 0.90 | 5mCpG detection |
| PacBio ccsmeth [45] | AUC | 0.97 | 5mCpG detection |
Applications in Genomic Contexts
Performance Across Genomic Features
Each technology demonstrates distinct strengths across different genomic contexts:
- GC-Rich Regions: EM-seq provides more uniform coverage in GC-rich regions compared to WGBS, which suffers from GC bias [42] [41]. ONT and PacBio also perform well in these regions without significant bias [42].
- Complex and Repetitive Regions: Long-read technologies excel in complex genomic regions, including repetitive elements and segmental duplications, where short-read technologies often fail [44] [45]. ONT sequencing enables methylation detection in challenging genomic regions that are inaccessible to other methods [44].
- Regulatory Elements: EM-seq shows enhanced detection of methylation in regulatory elements, capturing more CpGs within genomic features compared to bisulfite methods [41]. EPIC arrays target specific regulatory elements but are limited to predefined sites [42] [44].
- Tissue-Specific Applications: Studies comparing heart and liver tissues across multiple vertebrate species have demonstrated conserved tissue-specific methylation patterns detectable across technologies [46].
Methylation Heterogeneity Analysis
Methylation heterogeneity, representing cell-to-cell variation in methylation patterns, can be assessed using both short-read and long-read technologies. Computational methods like MeH have been developed to estimate genome-wide DNA methylation heterogeneity from pooled cell data [47]. Long-read technologies provide inherent advantages for heterogeneity studies as they can capture phased methylation patterns across long DNA molecules, enabling analysis of haplotype-specific methylation [45].
Practical Implementation Considerations
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for DNA Methylation Analysis
| Reagent/Resource | Function | Technology |
|---|---|---|
| TET2 Enzyme | Oxidizes 5mC to 5caC | EM-seq [41] |
| APOBEC3A | Deaminates unmodified cytosines | EM-seq [41] |
| T4-BGT | Glucosylates 5hmC for protection | EM-seq [41] |
| Nanopolish | Detects methylation from raw signals | ONT [43] |
| ccsmeth | Detects 5mCpGs from kinetic features | PacBio CCS [45] |
| ccsmethphase | Haplotype-aware methylation detection | PacBio CCS [45] |
| Methylation Callers | (e.g., Megalodon, DeepSignal) | ONT [45] |
| RRBS Kit | Reduced representation bisulfite sequencing | Traditional [46] |
Method Selection Guidelines
Choosing the appropriate methylation profiling technology depends on research goals, sample characteristics, and resource constraints:
- EM-seq is recommended for: (1) Low-input DNA samples (pg-ng range) [41], (2) Studies requiring minimal DNA degradation, (3) Projects needing accurate quantification across GC-rich regions, and (4) Simultaneous detection of 5mC and 5hmC [42] [41].
- Long-read sequencing (ONT/PacBio) is ideal for: (1) Methylation phasing and haplotype-aware analysis [45], (2) Complex genomic regions with repeats, (3) Integrated variant and methylation detection, and (4) De novo methylation profiling without reference bias [44] [43].
- WGBS remains suitable for: (1) Well-funded projects with sufficient DNA quantity and quality, (2) Established pipelines requiring extensive comparable datasets, and (3) Applications where cost is a primary constraint [42].
- EPIC arrays are practical for: (1) Large cohort studies with limited budgets, (2) Clinical applications targeting known regulatory CpGs, and (3) Laboratories with limited bioinformatics resources [44].
EM-seq and long-read sequencing technologies have significantly expanded the capabilities for DNA methylation analysis, each offering distinct advantages over traditional bisulfite-based methods. EM-seq provides superior performance for low-input samples and GC-rich regions through its gentle enzymatic conversion process, while long-read technologies enable methylation phasing, profiling of complex genomic regions, and direct detection without conversion. The choice between these technologies should be guided by specific research objectives, sample characteristics, and analytical requirements. As these technologies continue to evolve, they are poised to overcome current limitations in cost and complexity, further expanding our understanding of DNA methylation in gene regulation, development, and disease.
DNA methylation, the process of adding a methyl group to a cytosine base in a CpG dinucleotide, is a fundamental epigenetic mechanism that regulates gene expression without altering the underlying DNA sequence [48]. In cancer and other diseases, global DNA methylation patterns are frequently disrupted, with simultaneous genome-wide hypomethylation and site-specific hypermethylation of tumor suppressor gene promoters [49] [50]. These aberrant methylation patterns emerge early in tumorigenesis and remain stable throughout disease progression, making them exceptionally valuable as biomarkers for early detection, diagnosis, and monitoring [48]. The stability of the DNA molecule itself, coupled with the rapid clearance of cell-free DNA (cfDNA) from circulation (with half-lives ranging from minutes to a few hours), further enhances the utility of DNA methylation biomarkers in liquid biopsies [48].
The selection of an appropriate DNA methylation analysis method is paramount to the success of any biomedical research or clinical diagnostic endeavor. The choice fundamentally hinges on the specific application and revolves around the core distinction between locus-specific methods, which target predefined genomic regions, and genome-wide approaches, which provide comprehensive epigenetic profiling [49] [50]. Locus-specific methods offer high sensitivity and are tailored for validating known biomarkers or applications in clinical diagnostics where throughput and cost are considerations. In contrast, genome-wide methods are discovery-oriented, enabling the unbiased identification of novel methylation markers across the entire epigenome [51]. This guide provides a structured comparison of DNA methylation technologies, matching their capabilities to key applications in biomarker validation, clinical diagnostics, and novel discovery.
Technology Comparison: Methodologies and Workflows
Established DNA Methylation Detection Methods
DNA methylation analysis technologies can be broadly categorized into established and emerging methods. Established techniques include both non-next-generation sequencing (non-NGS) and NGS-based platforms [50].
Locus-Specific Methods (Non-NGS-based) are typically used for targeted analysis and biomarker validation. These include:
- Methylation-Specific PCR (MSP) and quantitative MSP (qMSP): These PCR-based methods allow for cost-effective, sensitive detection of hypermethylated CpG sites in known genes but are limited to predefined CpG sites [50].
- Droplet Digital PCR (ddPCR): This method enhances quantification accuracy for low-abundance methylation signals in liquid biopsies, offering absolute quantification without the need for standard curves [49] [50].
- Pyrosequencing: A sequencing-by-synthesis method that provides real-time, quantitative methylation analysis across multiple consecutive CpG sites within a short amplicon, making it highly valuable for biomarker validation [50] [31]. It is considered one of the most robust methods for quantitative methylation analysis [52] [53].
- Methylation-Specific High-Resolution Melting (MS-HRM): A post-PCR method that analyzes the melting behavior of bisulfite-converted DNA to determine methylation status. It is a simple, quick, and accurate method that does not require probes or sequencing [31].
Epigenome-Wide and Targeted Sequencing Methods (NGS-based) have transformed methylation profiling by providing single-base resolution:
- Whole-Genome Bisulfite Sequencing (WGBS): Considered the gold standard for comprehensive, unbiased methylome-wide coverage, WGBS provides single-base resolution across the entire genome. However, its high cost, substantial DNA input requirements, and computational complexity limit its routine use [49] [50].
- Reduced Representation Bisulfite Sequencing (RRBS): This method lowers costs by selectively targeting CpG-rich regions, particularly promoters and enhancers, using restriction enzyme digestion. However, this approach results in incomplete genome coverage [49] [50].
- Infinium Methylation BeadChips (e.g., EPIC array): These microarrays enable high-throughput epigenetic profiling of up to 930,000 predefined CpG sites, facilitating large-scale epigenome-wide association studies at a relatively low cost per sample [49] [50].
Emerging Technologies for Enhanced Detection
Recent technological advances are addressing limitations of established methods, particularly for liquid biopsy applications:
- Bisulfite-Free Sequencing Methods: Techniques such as Enzymatic Methylation Sequencing (EM-seq) and Tet-Assisted Pyridine Borane Sequencing (TAPS) improve DNA integrity preservation and sequencing efficiency by avoiding the harsh bisulfite conversion process, which causes DNA fragmentation [49] [50].
- Direct Long-Read Sequencing: Technologies including Single-Molecule Real-Time Sequencing (SMRT-seq) and Oxford Nanopore sequencing eliminate the need for bisulfite treatment altogether, allowing for direct detection of base modifications in native DNA molecules. These approaches provide long-read sequencing capabilities that enhance the resolution of methylation patterns in fragmented circulating tumor DNA [49] [50].
- Low-Input and Low-Pass WGBS: Adapted versions of WGBS have been developed to work with as little as 1 ng of cell-free DNA, significantly enhancing feasibility for liquid biopsy applications where sample material is limited [50].
Comparative Performance Analysis
Table 1: Quantitative Comparison of DNA Methylation Analysis Methods
| Method | Technology Type | Coverage | DNA Input | CpG Resolution | Cost | Best Suited Application |
|---|---|---|---|---|---|---|
| Pyrosequencing | Locus-specific | Targeted regions | ≥20 ng | Single-base | $ | Clinical validation, biomarker verification |
| qMSP/MethyLight | Locus-specific | Targeted CpG sites | Low input | Site-specific | $ | High-throughput clinical screening |
| MS-HRM | Locus-specific | Targeted regions | Low input | Regional | $ | Rapid screening, clinical validation |
| Infinium BeadChip | Genome-wide | 930,000 CpG sites | ≥250 ng | Single-CpG | $$ | Large-scale association studies |
| RRBS | Genome-wide | CpG-rich regions | ≥30 ng | Single-base | $$$ | Discovery in functional genomic regions |
| WGBS | Genome-wide | All CpGs | ≥100 ng | Single-base | $$$$ | Comprehensive discovery, reference maps |
| EM-seq/TAPS | Genome-wide | All CpGs | ≥10-100 ng | Single-base | $$$$ | Discovery with enhanced DNA recovery |
Table 2: Benchmarking Performance Across Applications (Data from [52] [53])
| Method | Accuracy | Sensitivity for Low Input | Discriminatory Power | Ease of Clinical Implementation |
|---|---|---|---|---|
| Amplicon Bisulfite Sequencing | High | High | High | Moderate |
| Bisulfite Pyrosequencing | High | High | High | High |
| Methylation-Specific PCR | Moderate | High | Moderate | High |
| Methylation-Specific HRM | High | High | High | High |
| Infinium BeadChip | High | Moderate | High | Moderate |
| RRBS | High | Moderate | High | Low |
| MeDIP-seq | Moderate | Moderate | Moderate | Low |
Matching Methods to Research and Clinical Applications
Biomarker Validation: Prioritizing Precision and Reproducibility
In the biomarker validation pipeline, the emphasis shifts from discovery to rigorous verification of candidate markers. This phase requires methods that deliver high quantitative accuracy, reproducibility, and sensitivity for detecting subtle methylation changes, often in limited clinical samples.
Recommended Methods:
- Bisulfite Pyrosequencing: This method excels in validation studies due to its quantitative precision across multiple consecutive CpG sites, robust performance in interlaboratory comparisons, and reliability with DNA from various sources including formalin-fixed paraffin-embedded (FFPE) tissues [52] [53] [31]. Pyrosequencing provides sequence information immediately adjacent to each CpG site, confirming the assay's specificity and providing internal quality control.
- Methylation-Specific High-Resolution Melting (MS-HRM): MS-HRM offers a rapid, cost-effective validation solution that requires no specialized probes or sequencing. The method is highly accurate for classifying methylation levels and can be performed on standard real-time PCR instruments [31].
- Droplet Digital PCR (ddPCR): For liquid biopsy applications where detecting rare methylated alleles in a background of normal DNA is crucial, ddPCR provides absolute quantification and exceptional sensitivity, making it ideal for validating biomarkers in circulating tumor DNA [49] [50].
Validation Workflow Considerations: The validation phase often begins with testing candidate biomarkers in larger sample sets (typically 100-500 samples) using targeted methods like pyrosequencing or MS-HRM [31]. Promising markers then advance to clinical validation in even larger cohorts, where methods like qMSP or ddPCR may be implemented for higher throughput [48]. This structured approach ensures that only the most robust biomarkers progress to clinical assay development.
Clinical Diagnostics: Implementing Robust and Scalable Assays
Clinical diagnostics demands methods that are not only accurate but also practical for routine use. Key considerations include turnaround time, cost-effectiveness, ease of implementation in clinical laboratories, and compatibility with standard workflows.
Recommended Methods:
- Quantitative Methylation-Specific PCR (qMSP): This method dominates molecular diagnostics due to its rapid turnaround time, familiar technology platform for clinical laboratories, and minimal DNA requirements. FDA-approved tests like Epi proColon for colorectal cancer detection are based on qMSP technology [48].
- Pyrosequencing: For applications requiring quantitative results across multiple CpGs, pyrosequencing offers an excellent balance of quantitative precision and clinical feasibility. Its ability to detect methylation gradients is particularly valuable for prognostic stratification [52] [53].
- Digital PCR (dPCR/ddPCR): Emerging as a powerful tool for liquid biopsy applications, dPCR platforms provide the exceptional sensitivity needed to detect minimal residual disease or early-stage cancers when the tumor DNA fraction in blood is very low [49] [50].
Implementation Framework: Successful clinical implementation requires rigorous assay optimization, establishment of clinically relevant cut-off values, and thorough validation in the intended-use population [48]. Clinical-grade assays must demonstrate analytical validity (accuracy, precision, sensitivity, specificity), clinical validity (ability to correctly identify the clinical condition), and clinical utility (improvement in patient outcomes) [48].
Novel Biomarker Discovery: Unleashing the Power of Unbiased Screening
The discovery phase aims to identify novel methylation markers without preconceived notions of their genomic location. This requires comprehensive, unbiased approaches that survey methylation patterns across the entire genome or at least its most informative regions.
Recommended Methods:
- Infinium MethylationEPIC BeadChip: For large-scale epigenome-wide association studies (EWAS), the EPIC array provides an optimal balance of comprehensive coverage (over 930,000 CpG sites) and cost-effectiveness. Its standardized workflow enables high-throughput profiling of thousands of samples, facilitating robust statistical analysis [49] [50].
- Reduced Representation Bisulfite Sequencing (RRBS): This method captures approximately 10% of CpGs in the genome at single-base resolution, with preferential enrichment in CpG-rich regulatory regions such as gene promoters and enhancers. RRBS provides deeper coverage of these functionally relevant regions compared to microarrays at a reasonable cost [49] [51].
- Whole-Genome Bisulfite Sequencing (WGBS): As the most comprehensive discovery approach, WGBS Interrogates over 28 million CpG sites across the entire genome, enabling discovery in non-coding regulatory elements, intergenic regions, and repetitive elements that are poorly covered by other methods [49] [51].
Discovery Workflow Considerations: An effective discovery pipeline typically begins with genome-wide screening in carefully selected sample sets (typically 20-100 samples per group) using arrays or sequencing-based methods [54] [5]. Bioinformatic analysis then identifies differentially methylated regions (DMRs) between case and control groups. Integration with transcriptomic data can help prioritize functionally relevant methylation changes that correlate with gene expression [54]. Advanced computational methods like the EpiClass algorithm can further enhance discovery by leveraging methylation density distributions to improve classification performance, particularly in heterogeneous samples like liquid biopsies [55].
Integrated Experimental Design: From Discovery to Clinical Application
Decision Framework for Method Selection
The following workflow diagram illustrates the decision pathway for selecting appropriate DNA methylation analysis methods based on research goals and sample considerations:
Multi-Omics Integration and Advanced Bioinformatics
The integration of DNA methylation data with other molecular data types significantly enhances biomarker discovery and biological interpretation:
- Methylation-Expression Integration: Combining DNA methylation profiles with transcriptomic data allows researchers to identify functionally relevant epigenetic alterations. As demonstrated in rectal cancer research, this integrated approach can pinpoint genes with inverse correlation between promoter methylation and expression, highlighting potential therapeutic targets [54].
- Machine Learning Enhancement: Advanced computational algorithms are increasingly applied to DNA methylation data to identify subtle cancer-specific patterns and improve classification accuracy. Methods like EpiClass leverage methylation density distributions to enhance detection sensitivity in challenging samples like liquid biopsies [55].
- Multi-Cancer Biomarker Discovery: Integrative bioinformatics approaches that combine methylation profiling with comorbidity patterns and gene ontology analysis can identify pan-cancer biomarkers with utility across multiple cancer types, as evidenced by the identification of ALX3, NPTX2, and TRIM58 for low-survival-rate cancers [5].
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Materials for DNA Methylation Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Sodium Bisulfite | Chemical conversion of unmethylated cytosines to uracils | Core reagent for most protocols; modern kits reduce DNA degradation |
| Methylation-Specific Restriction Enzymes (e.g., HpaII) | Differential digestion based on methylation status | Used in MSRE approaches; no bisulfite conversion needed |
| 5-methylcytosine Antibodies | Immunoprecipitation of methylated DNA | Used in MeDIP-seq; enriches for methylated regions |
| MBD Capture Proteins | Affinity purification of methylated DNA | Used in MethylCap-seq; captures DNA based on methylation density |
| Bisulfite Conversion Kits | Complete cytosine conversion while preserving methylated cytosines | Critical step for bisulfite-based methods; commercial kits offer improved efficiency |
| PCR Primers for Bisulfite-Converted DNA | Amplification of target regions after conversion | Must be designed for sequence after bisulfite treatment; avoid CpG sites in primers |
| Probes for Methylation-Specific PCR | Detection of specific methylation patterns | Fluorescent probes (TaqMan) or SYBR Green for real-time detection |
| Biotinylated Primers | Capture of PCR products for pyrosequencing | Required for preparation of single-stranded DNA template in pyrosequencing |
The selection of an appropriate DNA methylation analysis method requires careful consideration of the research objective, sample characteristics, and practical constraints. Locus-specific methods like pyrosequencing, MS-HRM, and qMSP offer precision, sensitivity, and practical advantages for biomarker validation and clinical diagnostics. In contrast, genome-wide approaches including Infinium BeadChips, RRBS, and WGBS provide the comprehensive profiling needed for novel discovery. Emerging bisulfite-free sequencing technologies promise to overcome historical limitations in DNA degradation while advanced computational methods enhance our ability to extract biological insights from complex methylation data. By strategically matching methods to applications across the research-to-clinical continuum, researchers can maximize the tremendous potential of DNA methylation as a biomarker for disease detection, monitoring, and personalized treatment.
Navigating Practical Challenges in DNA Methylation Analysis
The analysis of DNA methylation patterns provides powerful insights into gene regulation, cellular identity, and disease mechanisms. However, the reliability of this analysis is fundamentally constrained by sample quality and quantity, presenting significant challenges for researchers working with formalin-fixed paraffin-embedded (FFPE) tissues, cell-free DNA (cfDNA), and other low-input samples. The choice between locus-specific and genome-wide DNA methylation analysis methods must be carefully considered in the context of these sample limitations. While genome-wide approaches like whole-genome bisulfite sequencing (WGBS) offer comprehensive coverage, they typically require substantial DNA input and high-quality samples. In contrast, locus-specific methods often demonstrate superior performance with degraded or limited material, making them indispensable for many clinical and archival sample applications [53]. This guide objectively compares the performance of various DNA methylation analysis technologies across challenging sample types, supported by experimental data to inform method selection for research and diagnostic applications.
Technology Landscape: Methods for DNA Methylation Analysis
Core Technological Principles
DNA methylation analysis methods are built upon four fundamental principles for differentiating methylation states: (1) methylation-sensitive/dependent restriction enzymes, (2) antibody or methyl-binding protein-based enrichment, (3) chemical or enzymatic conversion, and (4) direct sequence readout without conversion [56] [8]. Bisulfite conversion remains the most widely adopted approach, chemically deaminating unmethylated cytosines to uracils while leaving methylated cytosines unchanged, thereby creating sequence differences that correspond to methylation states [44]. Second-generation sequencing has largely replaced microarrays as the primary readout platform, though arrays remain popular for large-scale studies due to their cost-effectiveness and standardized processing [56] [8].
Table 1: Core DNA Methylation Analysis Technologies and Their Characteristics
| Technology | Principle | Resolution | Primary Applications | Key Advantages |
|---|---|---|---|---|
| Whole-Genome Bisulfite Sequencing (WGBS) | Bisulfite conversion + NGS | Single-base | Genome-wide discovery | Comprehensive coverage; ~80% of CpGs [44] |
| Infinium MethylationEPIC Array | BeadChip microarray | Predefined CpG sites | Large cohort studies | Cost-effective; standardized processing [44] |
| Enzymatic Methyl-Seq (EM-seq) | Enzymatic conversion + NGS | Single-base | Genome-wide with less damage | Preserves DNA integrity; improved CpG detection [44] |
| Nanopore Sequencing | Direct electrical readout | Single-base | Long-range methylation | No conversion needed; long reads [44] |
| Amplicon Bisulfite Sequencing | Target amplification + NGS | Single-base | Locus-specific validation | Excellent for low-input; high sensitivity [53] |
| Bisulfite Pyrosequencing | Sequencing by synthesis | Single-base | Locus-specific validation | Quantitative; rapid turnaround [53] |
Emerging Technologies and Innovations
Recent methodological advances aim to overcome limitations associated with traditional bisulfite treatment. Enzymatic methyl-sequencing (EM-seq) uses the TET2 enzyme and T4-β-glucosyltransferase to convert and protect methylated cytosines, offering a gentler alternative that preserves DNA integrity and reduces sequencing bias [44]. Third-generation sequencing technologies, particularly Oxford Nanopore Technologies (ONT), enable direct detection of DNA methylation without chemical or enzymatic treatments by measuring electrical current deviations as DNA passes through protein nanopores [44]. These emerging methods show particular promise for analyzing challenging samples, as they minimize DNA damage and can handle more degraded material.
Sample-Specific Challenges and Method Performance
Formalin-Fixed Paraffin-Embedded (FFPE) Samples
DNA Quality Challenges in FFPE Material
FFPE samples represent an invaluable resource for retrospective studies, but formalin fixation causes substantial DNA damage through cross-linking, fragmentation, and generation of apurinic/apyrimidinic sites [57]. This degradation poses significant challenges for whole-genome amplification (WGA), an integral step in many genome-wide methylation assays including the Infinium platform [57]. Without modification, standard Infinium chemistry fails with fragmented FFPE DNA, necessitating specialized protocols for reliable analysis [57].
Performance Assessment of FFPE-Compatible Methods
Several studies have systematically evaluated methylation analysis methods using paired FFPE and fresh-frozen (FF) samples. A study comparing Infinium Methylation27K data from paired FF and FFPE colorectal tissues found that while overall correlation of β-values was reasonable, tissue storage type (FF vs. FFPE) represented the most significant source of variation rather than tissue type (normal vs. tumor) [57]. Critically, the concordance of differentially methylated loci (DML) detected between FF and FFPE DNA was suboptimal – when comparing the top 50 DML between tumor and normal tissue in both FF and FFPE samples, only 7 were common [57]. Similarly, less than 10% of individual loci showed strong correlation (r ≥ 0.6) across patients [57].
A methodological study of breast tumors using the Infinium HumanMethylation450K platform demonstrated better performance, with greater than 84% of the top 100 loci previously shown to differentiate ER+ and ER- tumors in FF tissues also identified as differentially methylated in FFPE samples [58]. Correlation analysis revealed strong agreement between FF and FFPE samples (mean ρ > 0.95), though detection rates and data quality were dependent on pre-analytical factors [58]. These findings support an emerging consensus that the 450K/EPIC platform can be employed to investigate epigenetics in archival FFPE tissues, albeit with careful quality control.
Table 2: Performance Metrics for DNA Methylation Analysis of FFPE Samples
| Study | Platform | Sample Type | Correlation (FF vs. FFPE) | DML Concordance | Key Limitations |
|---|---|---|---|---|---|
| Espinal et al. (2017) [58] | Infinium 450K | Breast tumors | Mean ρ > 0.95 | >84% of top ER-status loci | Sample-dependent variability |
| Kandimalla et al. (2012) [57] | Infinium 27K | Colorectal tissues | Comparable to previous studies | Poor (7/50 top DML common) | Ligation repair needed |
| Kandimalla et al. (2012) [57] | Modified Infinium (ligation) | Colorectal tissues | Reasonable for detected loci | Suboptimal for DML | Much fewer detected loci vs. FF |
Cell-Free DNA and Liquid Biopsies
Unique Characteristics of Cell-Free DNA
Cell-free DNA from liquid biopsies presents distinct challenges for methylation analysis, including low abundance of target molecules, short fragment length, and the presence of background methylation from normal cells. Genome-wide methylation analysis of cfDNA has revealed that fragments are enriched in coding and intergenic regions while being depleted from gene promoter regions [59]. The amount of cfDNA fragments shows a negative correlation with CpG density and GC content, making certain genomic regions more accessible for analysis than others [59].
Advanced Analysis Methods for cfDNA
The EpiClass algorithm represents an innovative approach to address methylation heterogeneity in cfDNA. This classification method leverages statistical differences in single-molecule methylation density distributions to optimize sample classification, particularly benefiting challenging samples like liquid biopsies [13]. When applied to ovarian carcinoma detection using ZNF154 methylation in plasma samples, EpiClass achieved 91.7% sensitivity and 100% specificity in an independent validation cohort, outperforming standard CA-125 measurement [13].
A prospective multi-center study on early-stage breast cancer detection demonstrated that cfDNA methylation markers significantly improved diagnostic accuracy for BI-RADS category 4 patients, with area under the curve (AUC) increasing from 0.78-0.79 with imaging alone to 0.93-0.94 when combined with cfDNA methylation analysis [59]. The study utilized whole-genome bisulfite sequencing of cfDNA from 203 patients and identified ten optimal hypomethylated regions as biomarkers, mostly located in intergenic regions [59].
Low-Input and Degraded Samples
For low-input samples, method selection must balance DNA requirements with information needs. A comprehensive benchmarking study comparing DNA methylation assays for biomarker development found that amplicon bisulfite sequencing and bisulfite pyrosequencing showed the best all-round performance across multiple sample types, including low-input scenarios [53]. Enzymatic conversion methods like EM-seq offer particular advantages for low-input samples, as they can handle lower DNA amounts while preserving molecular integrity [44].
Experimental Protocols for Challenging Samples
Ligation-Based Protocol for FFPE DNA
The Thirlwell modified protocol enables Infinium methylation analysis of FFPE samples through DNA repair prior to bisulfite conversion [57]:
- DNA Extraction: Extract DNA from FFPE tissue using the Puregene Core kit A (Qiagen) or similar, with RNase treatment to remove RNA contamination.
- Quality Assessment: Assess DNA concentration by Nanodrop and integrity by Agilent Bioanalyzer 2100 using the DNA 12000 kit.
- Ligation-Based Repair: Ligate 2μg of FFPE DNA using Thirlwell's ligation-based repair method to reconstruct fragmented DNA.
- Bisulfite Conversion: Perform bisulfite conversion using the EZ DNA methylation kit (Zymo Research) following manufacturer's recommendations.
- Array Processing: Process samples on the Infinium Methylation array using automated sample processing (e.g., Tecan Evo robot) and scan with a BeadArray reader.
- Quality Control: Ensure staining intensity >15,000, clear clustering for hybridization probes, target removal intensity <400, and satisfactory bisulfite conversion.
Whole-Genome Bisulfite Sequencing for Cell-Free DNA
A protocol for genome-wide cfDNA methylation analysis from blood-based liquid biopsies [59]:
- Plasma Collection and cfDNA Extraction: Collect median of 3mL plasma from participants before surgery or biopsy. Extract cfDNA using specialized kits preserving short fragments.
- Size Selection and Library Preparation: Retain fragments lower than 500bp during bead-based library purification to remove genomic DNA contamination. Use Agilent 2100 Bioanalyzer to assess size distribution (typical peak at 167bp).
- Whole-Genome Bisulfite Sequencing: Perform WGBS with appropriate coverage (study achieved 4.0Tb total data, ~88% reference genome coverage, 11.2× average depth).
- Bioinformatic Processing: Identify differentially methylated regions (DMRs) between malignant and benign samples. Select hypomethylated DMRs as candidate biomarkers due to their higher cfDNA fragment count.
- Marker Validation: Compute cfDNA malignant ratios for hypo-DMRs and select optimal markers using discovery cohort, then validate in independent cohort.
EpiClass Methodology for Methylation Density Analysis
The EpiClass protocol for leveraging methylation density in heterogeneous samples [13]:
- Data Generation: Generate epiallelic methylation density distributions using any method that provides single-molecule resolution (RRBS, WGBS, or digital techniques like DREAMing).
- Tabulate Methylation Density: For each detected epiallelic copy of the target locus, count the number of methylated CpG sites.
- Calculate Epiallelic Fractions: Determine the relative abundance of epialleles at each methylation density level.
- Identify Optimal Cutoffs: Use the algorithmic procedure to identify methylation density and epiallelic fraction cutoffs that optimize classification of case and control samples.
- Validation: Confirm classifier performance in independent sample cohorts, comparing against traditional metrics like mean methylation levels.
Decision Framework and Comparative Analysis
The selection of appropriate DNA methylation analysis methods depends on multiple factors including sample type, quality, quantity, and research objectives. The following diagram illustrates the method selection workflow for different sample scenarios:
Diagram 1: DNA methylation analysis method selection workflow for different sample types
Performance Comparison Across Sample Types
Table 3: Comprehensive Performance Comparison of DNA Methylation Analysis Methods
| Method | Optimal Sample Type | Minimum Input | CpG Coverage | Cost Considerations | Technical Challenges |
|---|---|---|---|---|---|
| WGBS | High-quality DNA | 1μg [44] | ~80% of CpGs [44] | High sequencing costs | Bisulfite-induced degradation; data complexity |
| EPIC Array | FFPE, large cohorts | 500ng [44] | 935,000 sites | Moderate per sample | Predefined coverage; limited discovery |
| EM-seq | Low-input, degraded | Lower than WGBS [44] | Comparable to WGBS | Moderate-high | Protocol complexity |
| Nanopore | Long-range methylation | ~1μg of 8kb fragments [44] | Genome-wide | Instrument cost | High DNA input requirements |
| AmpliconBS | Low-input, FFPE, cfDNA | As low as 1ng | Locus-specific | Low-moderate | Multiplexing limitations |
| Pyroseq | Clinical validation | 10-50ng | Locus-specific | Low | Limited genomic coverage |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents and Their Applications in DNA Methylation Analysis
| Reagent/Kit | Primary Function | Sample Applications | Key Considerations |
|---|---|---|---|
| EZ DNA Methylation Kit (Zymo Research) | Bisulfite conversion | All bisulfite-based methods | Complete conversion critical for accuracy [57] [44] |
| Puregene Core Kit A (Qiagen) | DNA extraction from FFPE | FFPE samples | Includes RNase treatment; compatible with degraded DNA [57] |
| Agilent Bioanalyzer DNA Kits | DNA quality assessment | All sample types | Essential for evaluating fragmentation; 12000 kit for FFPE [57] |
| Infinium MethylationEPIC v2.0 BeadChip | Genome-wide methylation | Large cohort studies | Covers >935,000 sites; optimized for clinical samples [44] |
| Nanobind Tissue Big DNA Kit | High-quality DNA extraction | Fresh-frozen tissues | Optimal for WGBS and other sequencing approaches [44] |
| TET2 Enzyme + T4-BGT | Enzymatic conversion | EM-seq protocols | Gentler alternative to bisulfite; preserves DNA integrity [44] |
The comparative analysis of DNA methylation methods across challenging sample types reveals that method selection must be guided by sample characteristics and research objectives. For FFPE samples, ligation-based repair methods followed by array-based analysis or targeted bisulfite sequencing provide the most reliable results, though with recognized limitations in DML concordance compared to fresh-frozen material [57] [58]. For cell-free DNA applications, WGBS with specialized bioinformatic approaches like methylation density analysis offers superior performance for cancer detection, particularly when combined with traditional diagnostic methods [59] [13].
Emerging technologies including enzymatic conversion methods and direct sequencing approaches promise to overcome many current limitations, particularly for degraded and low-input samples [44]. The ongoing development of specialized algorithms to address methylation heterogeneity, such as EpiClass, further enhances our ability to extract meaningful biological signals from challenging samples [13]. As these technologies mature and become more accessible, they will undoubtedly expand the research and clinical applications of DNA methylation analysis across all sample types, ultimately enabling more comprehensive and reliable epigenetic profiling in both basic research and translational applications.
DNA methylation, a fundamental epigenetic modification, is pivotal for gene regulation, cellular differentiation, and embryonic development [60]. Its analysis is essential for understanding biological processes and disease mechanisms, with bisulfite conversion (BC) serving as the decades-long gold standard method [61] [20]. This chemical process selectively deaminates unmethylated cytosines to uracils, while methylated cytosines remain intact, creating sequence differences that reveal methylation status [61]. Despite its widespread adoption, BC presents significant challenges, including substantial DNA degradation, incomplete conversion leading to false positives, and reduced sequence complexity, which are particularly problematic for low-quality and low-quantity DNA samples such as cell-free DNA (cfDNA) and forensic samples [61] [62] [20]. This guide objectively compares bisulfite-based methods with emerging enzymatic alternatives, providing researchers with experimental data and protocols to navigate the critical trade-offs between conversion completeness and DNA preservation in both locus-specific and genome-wide methylation analysis.
Comparative Performance Analysis of DNA Conversion Methods
Quantitative Performance Metrics
The following table summarizes key performance indicators for major DNA conversion methods, synthesized from recent comparative studies:
Table 1: Performance Comparison of DNA Methylation Conversion Methods
| Method | Conversion Efficiency | DNA Recovery | DNA Fragmentation | Optimal Input DNA | Background Noise | Best Application Context |
|---|---|---|---|---|---|---|
| Conventional Bisulfite (CBS) | >99.5% [63] | Overestimated (130%) [61] [20] | High (14.4±1.2) [61] [20] | 5 ng [61] [20] | <0.5% [62] | Standard genomic DNA with sufficient quantity |
| Enzymatic Conversion (EC) | Similar to BC [61] [20] | Low (40%) [61] [20] | Low-Medium (3.3±0.4) [61] [20] | 10 ng [61] [20] | >1% at low inputs [62] | Degraded DNA, cell-free DNA, long-range methylation |
| Ultra-Mild Bisulfite (UMBS) | ~0.1% unconverted cytosines [62] | Higher than EM-seq and CBS [62] | Significantly reduced vs. CBS [62] | As low as 10 pg [62] | ~0.1% [62] | Low-input cfDNA, clinical biomarker detection |
| Rapid Bisulfite (90°C) | >99.5% in 10 min [63] | ~65% for cfDNA [63] | Reduced vs. conventional [63] | Low cfDNA levels [63] | N/R | Time-sensitive clinical applications |
Table 2: Library Preparation and Sequencing Performance
| Method | Library Complexity | Insert Size | GC Bias | Protocol Duration | Cost Considerations | Special Equipment |
|---|---|---|---|---|---|---|
| Conventional Bisulfite | Lower (high duplication rates) [62] | Shorter fragments [62] | Substantial GC bias [62] | 16+ hours (standard) [20] | Lower reagent cost [62] | Standard thermal cycler |
| Enzymatic Conversion | Higher than CBS [62] | Longer, comparable to UMBS [62] | Reduced GC bias [62] | 4.5 hours [20] | Higher reagent cost [62] | - |
| Ultra-Mild Bisulfite | High (lower duplication rates) [62] | Longer, comparable to EM-seq [62] | Improved uniformity [62] | 90 minutes [62] | Moderate | Standard thermal cycler |
Impact on Locus-Specific vs. Genome-Wide Analysis
The choice of conversion method differentially affects locus-specific and genome-wide applications:
Locus-Specific Analysis: BC's DNA fragmentation poses significant challenges for amplifying longer targets, while enzymatic conversion (EC) preserves DNA length, benefiting multi-locus panels [61]. Incomplete conversion in GC-rich regions affects primer binding specificity in both approaches [30].
Genome-Wide Analysis: BC reduces sequence complexity to a 3-letter genome, complicating alignment and increasing duplication rates [20]. EC and UMBS demonstrate improved coverage of GC-rich regulatory elements including promoters and CpG islands, providing more comprehensive methylome coverage [62].
Experimental Protocols for Method Evaluation
qBiCo Multiplex qPCR Assessment Protocol
The qPCR-based Bisulfite Conversion (qBiCo) quality control method enables standardized evaluation of conversion performance [20]:
Table 3: Research Reagent Solutions for qBiCo Assessment
| Reagent/Assay | Function | Target | Application in Quality Control |
|---|---|---|---|
| Genomic Assay | Detects DNA regardless of deamination | Multi-copy human L1 repetitive element (~200 copies) | Baseline DNA quantification |
| Converted Assay | Detects deaminated DNA | Converted version of L1 element | Conversion efficiency calculation |
| Short Assay | Quantifies converted DNA | Converted single-copy hTERT gene | Converted DNA concentration |
| Long Assay | Detects longer converted fragments | Converted longer version of single-copy TPT1 gene | DNA fragmentation assessment |
Procedure:
- Convert DNA samples using standardized protocols for each method
- Perform multiplex qPCR with all four assays simultaneously
- Calculate conversion efficiency: (1 - [Genomic/Converted]) × 100%
- Determine converted DNA concentration using Short assay standard curve
- Assess fragmentation: ratio of Long to Short assay signals
- Analyze data with efficiency thresholds: >99% conversion, <10% fragmentation ideal
Validation Parameters: Test repeatability (intra-run precision), reproducibility (inter-run, inter-operator), sensitivity (limit of detection), and robustness (protocol variations) [20].
Ultra-Mild Bisulfite Conversion Protocol
The UMBS-seq method optimizes bisulfite chemistry to minimize DNA damage [62]:
Reagent Formulation:
- 100 μL of 72% ammonium bisulfite
- 1 μL of 20 M KOH
- DNA protection buffer (component not specified)
Procedure:
- Mix DNA sample with optimized bisulfite formulation
- Incubate at 55°C for 90 minutes (significantly milder than conventional 16-18 hours at high temperatures)
- Include alkaline denaturation step to maintain DNA single-strandedness
- Purify using bead-based cleanup
- Proceed to library preparation
Validation: Complete conversion of model oligonucleotide while preserving 5mC integrity, with significantly reduced DNA damage compared to conventional bisulfite [62].
Enzymatic Conversion Workflow
The NEBNext Enzymatic Methyl-seq Conversion Module protocol [20]:
Procedure:
- Oxidation: Treat DNA with TET2 enzyme to convert 5mC to 5caC
- Glycosylation: Use T4-BGT to glucosylate 5hmC, protecting it from deamination
- Deamination: Treat with APOBEC to deaminate unmodified cytosines to dihydrouracil (DHU)
- Purification: Two bead-based cleanup steps (noted as tedious and time-consuming when performed manually)
- Library Preparation: DHU is read as thymine during PCR amplification
Key Considerations: Enzymatic instability and increased background at low inputs (exceeding 1% unconverted cytosines at lowest inputs) require careful quality control [62].
Method Workflow and DNA Integrity Visualization
The following diagram illustrates the procedural steps and DNA impact across the three main conversion technologies:
Advanced Applications and Research Context
Applications in Challenging Sample Types
Different conversion methods show varying performance with difficult samples:
Cell-Free DNA (cfDNA): UMBS and EC effectively preserve the characteristic cfDNA triple-peak profile after treatment, while conventional bisulfite causes significant fragmentation [62]. UMBS-seq consistently produces higher library yields and greater complexity than EM-seq across low input levels (5 ng to 10 pg) [62].
Formalin-Fixed Paraffin-Embedded (FFPE) Tissue: DNA from FFPE samples is already fragmented, making them particularly susceptible to additional degradation from harsh bisulfite treatment [62]. Enzymatic approaches show promise for these valuable clinical specimens.
Forensic and Ancient DNA: The low fragmentation characteristic of EC makes it suitable for forensic-type DNA and other degraded samples where template preservation is paramount [61] [20].
Integration with Broader Methylation Analysis Research
The conversion method selection directly impacts data quality in broader methylation research contexts:
Locus-Specific vs. Genome-Wide Trade-offs: Bisulfite conversion remains compatible with most established analysis platforms, including methylation-specific PCR, pyrosequencing, and the Illumina EPIC array [30] [20]. Enzymatic conversion shows strong concordance with whole-genome bisulfite sequencing (WGBS) but requires validation for targeted applications [30].
Emerging Technologies: Third-generation sequencing technologies like Oxford Nanopore Technologies (ONT) enable direct methylation detection without conversion, capturing certain loci uniquely and enabling methylation detection in challenging genomic regions, though with different performance characteristics [30].
The evolution of bisulfite conversion methodologies addresses the persistent challenge of balancing conversion completeness with DNA integrity. Conventional bisulfite conversion offers robust performance with standard DNA samples but shows limitations with delicate samples. Enzymatic conversion minimizes DNA damage but faces challenges with recovery efficiency and background noise at low inputs. Ultra-mild bisulfite methods represent a promising intermediate, leveraging the robustness of bisulfite chemistry while significantly reducing DNA damage.
For researchers selecting conversion methods, the decision matrix should prioritize:
- Sample Quality: Degraded samples benefit from enzymatic or ultra-mild bisulfite approaches
- Input Amount: Low-input applications (<10 ng) require careful optimization regardless of method
- Application Scope: Genome-wide studies benefit from higher complexity libraries, while locus-specific assays require optimized conversion in target regions
- Practical Considerations: Protocol duration, cost, and automation compatibility vary substantially between methods
This comparative analysis provides the experimental framework for researchers to implement and validate DNA methylation conversion methods appropriate for their specific research context, advancing the reliability of both locus-specific and genome-wide epigenetic studies.
The choice of an appropriate method for DNA methylation analysis is a critical first step in any epigenetics study, fundamentally shaping the experimental design, scope of biological questions that can be addressed, and the required bioinformatics pipeline. The field is broadly divided into locus-specific techniques, which investigate predefined genomic regions, and genome-wide approaches, which allow for the discovery of novel methylation patterns across the entire genome [24]. This guide provides an objective comparison of the performance of various wet-lab methods and their corresponding computational pipelines, framing them within the context of a broader thesis that contrasts the targeted power of locus-specific analysis with the discovery potential of genome-wide profiling. We summarize quantitative performance data and provide detailed experimental protocols to assist researchers, scientists, and drug development professionals in selecting the optimal path for their research.
Comparison of DNA Methylation Analysis Methods
The three principal experimental techniques for DNA methylation profiling are based on different underlying principles to distinguish methylated from unmethylated cytosines: bisulfite conversion, affinity enrichment, and restriction enzyme digestion [29] [64]. The choice among them depends on the biological question, required resolution, quantitative nature of the data needed, and available budget [29].
Table 1: Core Characteristics of Major DNA Methylation Analysis Methods
| Method | Principle | Resolution | Genome Coverage | Primary Application | Cost & Throughput |
|---|---|---|---|---|---|
| WGBS | Bisulfite Conversion | Single-base | ~50% of genome (≥2 CpG/100bp) [15] | Gold standard for base-resolution methylomes [29] | High cost, low to medium throughput |
| RRBS | Bisulfite Conversion + Enzymatic Digestion | Single-base | <20% of genome (>3 CpG/100bp) [15] | Cost-effective profiling of CpG-rich regions [11] | Medium cost, medium throughput |
| MeDIP-Seq | Affinity Enrichment (Antibody) | ~100-1000 bp | >95% of genome (<5 CpG/100bp) [15] | Global methylation profiling, low CpG density regions [15] | Medium cost, medium throughput |
| MRE-Seq | Restriction Enzyme Digestion | ~100-1000 bp | Limited to enzyme recognition sites [29] | Profiling unmethylated regions [29] | Medium cost, medium throughput |
| Methylation Arrays (e.g., Illumina Infinium) | Bisulfite Conversion + Probe Hybridization | Single-base (predefined sites) | ~1% of genome (predefined CpG sites) [29] | High-throughput population studies [29] | Low cost, high throughput |
Key Performance Metrics and Experimental Data
Different methods exhibit distinct biases and performance characteristics, which are crucial for data interpretation. A major factor is CpG density, which varies widely across the genome.
Table 2: Quantitative Performance Comparison of Genome-Wide Methods
| Method | CpG Density Bias | Typical Sequence Alignment Rate | Sensitivity to Small Methylation Changes | Ability to Detect 5hmC | DNA Input Requirements |
|---|---|---|---|---|---|
| WGBS | High-density bias (≥2 CpG/100bp) [15] | ~75% [15] | High (single-base resolution) | No (bisulfite cannot distinguish 5mC from 5hmC) [29] | High (micrograms) |
| RRBS | High-density bias (>3 CpG/100bp) [15] | ~75% [15] | High (single-base resolution) | No [29] | Low (nanograms) |
| MeDIP-Seq | Low-density bias (<5 CpG/100bp) [15] | >95% [15] | Low to Medium (region-based) | Yes (if using 5hmC-specific antibody) [29] | Medium (hundreds of nanograms) |
| MRE-Seq | Dependent on restriction site density [29] | High (standard DNA sequencing) | Medium (region-based) | No | Medium (hundreds of nanograms) |
The table above shows that the majority of a typical vertebrate genome (>90%) falls into the low CpG density category (1-3 CpGs/100 bp), with less than 10% in the high density (>5 CpGs/100 bp) category [15]. Consequently, no single technique can provide a complete 100% assessment of the genome-wide methylation landscape [15]. MeDIP-Seq covers the largest percentage of the genome (>95%) but is biased toward low-density regions, whereas RRBS and WGBS offer higher resolution but focus on a smaller, CpG-rich portion of the genome [15].
Experimental Protocols for Key Methods
Whole-Genome Bisulfite Sequencing (WGBS)
Principle: This gold-standard method uses sodium bisulfite to convert unmethylated cytosines to uracils (which are read as thymines in sequencing), while methylated cytosines remain unchanged [65]. The sequencing results are then compared to a reference genome to determine the methylation status of each cytosine at single-base resolution [15].
Detailed Protocol:
- DNA Extraction and Fragmentation: High-quality genomic DNA is extracted and randomly sheared via sonication to a size of a few hundred base pairs [15].
- Bisulfite Conversion: DNA is treated with sodium bisulfite. This step requires optimization of time and temperature to ensure complete conversion of unmethylated cytosines while minimizing DNA degradation [64].
- Library Preparation and PCR: Converted DNA fragments are equipped with sequencing adapters and amplified using PCR. Special care must be taken during this step, as bisulfite-treated DNA has reduced complexity, which can lead to biased amplification [15].
- Sequencing: Libraries are sequenced on a next-generation sequencing platform (e.g., Illumina) to achieve sufficient depth for confident methylation calling.
- Bioinformatics Analysis: This critical step involves:
- Quality Control: Assessing raw read quality using tools like FastQC.
- Adapter Trimming: Removing adapter sequences with tools like Trim Galore! or Cutadapt.
- Alignment: Mapping bisulfite-converted reads to a reference genome using specialized aligners like Bismark [15] or BS-Seeker2 [15], which account for C-to-T conversions.
- Methylation Calling: Quantifying the percentage of methylated reads at each CpG site to generate a genome-wide methylation map.
- Differential Methylation Analysis: Identifying statistically significant Differentially Methylated Regions (DMRs) or Positions (DMPs) between samples using tools like methylKit or DMReate.
Methylated DNA Immunoprecipitation Sequencing (MeDIP-Seq)
Principle: This method uses an antibody specific for 5-methylcytosine (5mC) to immunoprecipitate methylated DNA fragments, which are then sequenced [15] [64].
Detailed Protocol:
- DNA Extraction and Shearing: Genomic DNA is extracted and sonicated to produce small fragments (200-500 bp). The DNA is then denatured into single strands to allow efficient antibody binding [15].
- Immunoprecipitation: Single-stranded DNA is incubated with a 5mC-specific antibody. The antibody-DNA complexes are isolated using magnetic beads coated with Protein G [64].
- Washing and Elution: Beads are washed stringently to remove non-specifically bound DNA. The methylated DNA is then eluted from the beads.
- Library Preparation and Sequencing: The eluted DNA is used to construct a sequencing library, which is amplified and sequenced [15].
- Bioinformatics Analysis:
- Read Mapping: Standard alignment tools like BWA [15] or Bowtie [15] are used, as the DNA is not bisulfite-converted and the sequence is not altered.
- Peak Calling: Tools like MEDIPS or MeDIPS are used to identify genomic regions significantly enriched for reads (peaks), which represent methylated regions.
- Differential Enrichment Analysis: Comparing peak intensities between samples to identify differentially methylated regions. As this is an enrichment-based method, it provides relative, not absolute, quantification of methylation levels.
Reduced Representation Bisulfite Sequencing (RRBS)
Principle: RRBS uses a methylation-insensitive restriction enzyme (e.g., MspI) to digest genomic DNA, enriching for CpG-rich regions. The size-selected fragments are then subjected to bisulfite sequencing [11] [15].
Detailed Protocol:
- DNA Digestion: Genomic DNA is digested with MspI, which cuts at CCGG sites regardless of methylation status.
- Size Selection: Digested DNA fragments are size-selected (e.g., 40-220 bp) to enrich for CpG islands and promoter regions [29].
- Bisulfite Conversion and Library Prep: The size-selected fragments undergo bisulfite conversion and standard library preparation for sequencing [15].
- Sequencing and Analysis: Libraries are sequenced. The bioinformatics pipeline is similar to WGBS, using bisulfite-aware aligners (Bismark) for mapping and methylation calling [11]. Its main advantage is the reduced sequencing cost and depth required due to the enrichment of informative, CpG-rich regions.
Diagram 1: DNA Methylation Analysis Workflow Selection. The workflow begins with DNA extraction, followed by a critical choice of the primary method, which dictates the subsequent wet-lab and computational steps.
The Scientist's Toolkit: Essential Reagents and Computational Tools
Successful DNA methylation analysis relies on a combination of wet-lab reagents and bioinformatics software.
Table 3: Research Reagent Solutions and Computational Tools
| Category | Item / Tool Name | Function / Application |
|---|---|---|
| Wet-Lab Reagents | Sodium Bisulfite | Chemical conversion of unmethylated cytosine for WGBS/RRBS [64]. |
| Anti-5-Methylcytosine (5mC) Antibody | Immunoprecipitation of methylated DNA for MeDIP-Seq [15] [64]. | |
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII) | Selective digestion of unmethylated DNA for MRE-based methods [64]. | |
| Methylation-Insensitive Restriction Enzymes (e.g., MspI) | Digestion for RRBS to define genomic representation [15] [11]. | |
| Bioinformatics Pipelines & Tools | Bismark [15] [66] | A widely used aligner and methylation caller for bisulfite sequencing data (WGBS, RRBS). |
| BWA / Bowtie [15] | Standard read aligners used for non-bisulfite converted data (e.g., MeDIP-Seq). | |
| MethylKit | An R package for differential methylation analysis from high-throughput bisulfite sequencing. | |
| SeSAMe | A pipeline for processing and analyzing Illumina methylation array data. | |
| MEDIPS | An R package for analyzing enrichment-based data like MeDIP-Seq. | |
| ClubCpG, MethylPurify [66] | Tools for cell-type deconvolution from bulk sequencing methylation data. |
Diagram 2: Core Bioinformatics Pipeline. A generalized computational workflow for DNA methylation data, highlighting the bifurcation at the alignment stage based on the initial method used.
The selection of a DNA methylation analysis method and its corresponding bioinformatics pipeline is a fundamental decision that directly determines the outcome and validity of an epigenetic study. As the field advances, technologies like single-cell bisulfite sequencing and long-read nanopore sequencing, which allows for direct detection of modified bases, are emerging [8]. However, the core trade-offs between resolution, genome coverage, cost, and analytical complexity remain. This guide provides a framework for researchers to make an informed choice, balancing their specific biological question with practical experimental and computational constraints. No single method is universally superior; the optimal path is the one that most precisely and reliably addresses the hypothesis at hand.
The selection of an appropriate DNA methylation analysis method is a critical decision that directly impacts the validity and scope of research findings. The choice between locus-specific techniques and genome-wide approaches presents a fundamental trade-off between depth and breadth, with optimal selection being highly dependent on the specific biological context under investigation. This guide provides an objective comparison of current methodologies, focusing on their performance across three particularly challenging research scenarios: interrogating repetitive genomic regions, detecting non-CpG methylation, and resolving cellular heterogeneity. As the field of epigenetics continues to reveal the complex relationship between methylation patterns and gene regulation, understanding the technical capabilities and limitations of available methods becomes increasingly important for researchers, scientists, and drug development professionals.
Technical Comparison of DNA Methylation Analysis Methods
The following tables summarize the key characteristics and performance metrics of major DNA methylation analysis platforms, highlighting their suitability for addressing specific research challenges.
Table 1: Core Characteristics of Major DNA Methylation Analysis Methods
| Method | Resolution | Throughput | DNA Input | Primary Applications |
|---|---|---|---|---|
| Whole-Genome Bisulfite Sequencing (WGBS) | Single-base | Genome-wide | Moderate to High (~1 µg) | Reference methylomes, non-CpG methylation, discovery studies [36] [37] |
| Enzymatic Methyl-Sequencing (EM-seq) | Single-base | Genome-wide | Low to Moderate | Superior coverage uniformity, DNA integrity preservation, non-CpG methylation [36] [37] |
| Oxford Nanopore Technologies (ONT) | Single-base (long-read) | Genome-wide | High (~1 µg of 8 kb fragments) | Long-range phasing, repetitive regions, direct methylation detection [36] [67] [37] |
| Illumina MethylationEPIC Array | Single CpG site | Targeted (850,000-935,000 sites) | Low (500 ng) | Large cohort studies, biomarker validation, clinical applications [5] [37] [68] |
| Methylation-Specific PCR (MS-PCR) | Locus-specific | Low (single locus) | Low | Validation studies, diagnostic assays for specific loci [69] |
Table 2: Performance Comparison for Challenging Genomic Contexts
| Method | Repetitive Regions | Non-CpG Methylation | Cost & Practicality | Key Limitations |
|---|---|---|---|---|
| WGBS | Limited due to short reads | Yes, but with bisulfite-conversion biases [37] | Moderate to High cost; complex data analysis [36] | DNA degradation, GC bias, computational intensity [36] [37] |
| EM-seq | Improved coverage over WGBS | Yes, with less DNA damage [37] | Moderate cost; less DNA damage [36] | Emerging method, less established protocols |
| ONT | Excellent (long reads span repeats) | Yes, via direct detection [67] | Moderate cost; minimal sample prep [36] | Higher DNA input, lower per-base accuracy [37] |
| EPIC Array | Poor (limited probe design in repeats) | No (targets CpG sites only) [37] | Low cost; high throughput; standardized [5] [37] | Targeted nature, unable to discover novel sites |
| MS-PCR | Dependent on primer design | No (typically designed for CpG) | Low cost; fast; simple analysis [69] | Ultra-specific, pre-knowledge of target required |
Analysis of Method Performance in Specific Research Contexts
Interrogating Repetitive Genomic Regions
Repetitive elements and heterochromatic regions pose significant challenges due to their complex nature and the difficulty in mapping short sequencing reads. Long-read sequencing technologies, such as Oxford Nanopore Technologies (ONT), excel in this context by spanning entire repetitive elements, allowing for unambiguous methylation profiling. Studies have successfully utilized ONT to achieve high levels of methylation in long repetitive sequences in heterochromatin, which are often poorly assessed by short-read methods [67]. While WGBS and EM-seq theoretically cover these regions, their resolution is limited by mapping ambiguities. Microarray-based methods are the least effective, as their probe design is inherently limited in repetitive regions [37].
Detecting Non-CpG Methylation
Non-CpG methylation (occurring in CHH and CHG contexts, where H is A, C, or T) is an important regulatory mark in plants, embryonic stem cells, and mammalian brains. Its analysis requires methods with single-base resolution that do not presuppose a sequence context. WGBS, EM-seq, and ONT sequencing are all capable of genome-wide non-CpG methylation profiling [37]. EM-seq offers a distinct advantage for this application due to its enzymatic conversion chemistry, which results in less DNA degradation and more uniform coverage compared to the harsh bisulfite treatment used in WGBS [36] [37]. In contrast, the Illumina EPIC array and other locus-specific methods like MS-PCR are designed to target specific CpG sites and are generally not suitable for investigating non-CpG methylation.
Resolving Cellular Heterogeneity
Cellular heterogeneity in tissue samples can obscure meaningful methylation patterns, as signals represent an average across cell types. While the methods discussed above typically analyze bulk DNA, their data can be integrated with computational deconvolution approaches to infer cell-type-specific methylation patterns. For genome-wide methods like WGBS, EM-seq, and EPIC arrays, bioinformatic tools can leverage reference methylomes of pure cell types to estimate proportions and signals from mixed samples [5]. The EPIC array's cost-effectiveness makes it particularly suitable for large-scale studies requiring high sample throughput, such as those profiling hundreds of cell lines to establish reference databases [68]. True single-cell methylome analysis requires specialized protocols that build upon the principles of WGBS or EM-seq, but are beyond the scope of standard genome-wide methods.
Essential Research Reagent Solutions
The following reagents and kits are fundamental to the experimental workflows of the DNA methylation methods discussed.
Table 3: Key Research Reagents for DNA Methylation Analysis
| Reagent / Kit | Function | Primary Application |
|---|---|---|
| EZ DNA Methylation Kit (Zymo Research) | Bisulfite conversion of unmethylated cytosines | WGBS, Illumina EPIC Array, post-bisulfite library prep [37] |
| NEBNext EM-Seq Kit (NEB) | Enzymatic conversion of unmethylated cytosines | EM-seq library preparation [36] |
| Infinium MethylationEPIC BeadChip (Illumina) | Microarray for CpG methylation quantification | Targeted genome-wide methylation analysis [5] [37] [68] |
| Nanobind Tissue Big DNA Kit (Circulomics) | Extraction of high-molecular-weight DNA | Long-read sequencing (e.g., ONT) [37] |
| MethylMiner Kit (Invitrogen) | Enrichment of methylated DNA via MBD-protein | Methylated DNA enrichment for sequencing or arrays [69] |
| Methylation-Specific Primers | PCR amplification of specific methylated loci | Validation and locus-specific analysis (MS-PCR) [69] |
Experimental Workflow and Data Analysis
The typical workflows for the major genome-wide methods share common steps but diverge in their core conversion or detection chemistry, which directly impacts data quality and application suitability. The following diagram illustrates the key stages of WGBS, EM-seq, and ONT sequencing.
For researchers analyzing data from the Illumina EPIC array, a standard bioinformatics pipeline involves several key steps. Data from IDAT files is processed using R packages like minfi or ChAMP for initial quality control, which involves removing underperforming probes, such as those with a detection p-value > 0.01, as well as control, multihit, and SNP-containing probes [5] [37]. Normalization is then performed using methods like Beta-Mixture Quantile (BMIQ) normalization to correct for technical variations between probe types [5]. Finally, methylation levels are reported as β-values, calculated as the ratio of the methylated probe intensity to the sum of methylated and unmethylated probe intensities, providing a value between 0 (unmethylated) and 1 (fully methylated) for downstream statistical analysis [5] [37].
The optimal DNA methylation analysis method is intrinsically linked to the specific research question and biological context. For discovery-based studies requiring comprehensive coverage, including non-CpG methylation, EM-seq emerges as a robust alternative to WGBS, offering superior data uniformity and DNA preservation [36] [37]. When investigating complex genomic regions such as repeats, ONT's long-read technology provides an unparalleled advantage [36] [67]. Conversely, for large-scale cohort studies or clinical biomarker validation where cost and throughput are paramount, the targeted Illumina EPIC array remains a powerful and efficient tool [5] [68]. Locus-specific methods retain their crucial role for hypothesis-driven validation. As the epigenetic field advances, the strategic selection and application of these complementary technologies will be fundamental to elucidating the functional role of DNA methylation in health and disease.
Benchmarking Performance and Selecting the Right Tool
DNA methylation analysis has become a cornerstone of epigenetic research, playing a critical role in understanding gene regulation, development, and disease mechanisms. The field primarily utilizes two complementary approaches: locus-specific methods that provide deep insights into predefined genomic regions, and genome-wide techniques that offer unbiased discovery across the entire epigenome. For researchers and drug development professionals, selecting the appropriate methodological pathway involves careful consideration of a validation framework built upon four fundamental pillars: accuracy, sensitivity, reproducibility, and robustness. This guide provides a comprehensive comparison of current DNA methylation analysis technologies, presenting structured experimental data and protocols to inform methodological selection for specific research and clinical applications. As the field advances toward clinical translation, with DNA methylation-targeted drugs already approved for hematological malignancies and numerous biomarkers in development [70] [71], establishing rigorous validation parameters becomes increasingly crucial for reliable data generation and interpretation.
Comparative Performance of DNA Methylation Analysis Methods
The selection of an appropriate DNA methylation analysis method requires careful evaluation of performance characteristics relative to research objectives. The table below summarizes the key technical parameters and applications of major analysis platforms.
Table 1: Performance Comparison of DNA Methylation Analysis Methods
| Method | Resolution | Throughput | Accuracy | Sensitivity | Best Applications |
|---|---|---|---|---|---|
| Amplicon Bisulfite Sequencing | Single-base | Medium | High | High (down to 1-5% for variants) | Targeted validation, biomarker verification [9] |
| Bisulfite Pyrosequencing | Single-base | Medium | High | High (down to 5% methylation) | Quantitative targeted analysis, clinical diagnostics [31] [9] |
| MS-HRM | Region-based | High | Medium | Medium (down to 0.1-10% methylation) | Screening, sample stratification [31] |
| MSRE-qPCR | Site-specific | High | Low-Medium | Low (requires >2 sites/amplicon) | Rapid screening of specific CpGs [31] |
| RRBS | Partial genome | Medium | High | High | Cost-effective discovery, cell-free DNA [8] [11] |
| Methylation Arrays | Predefined sites | Very High | High | Medium | Epigenome-wide association studies, biobank studies [72] [73] |
| Whole Genome Bisulfite Sequencing | Base-level genome-wide | Low | High (Gold standard) | High | Comprehensive discovery, single-cell analysis [8] [72] |
| Nanopore Sequencing | Base-level genome-wide | Medium | Emerging | Emerging | Simultaneous genetic/epigenetic analysis, large structural variants [14] |
A community-wide benchmarking study evaluating 27 different assays across 18 laboratories demonstrated that amplicon bisulfite sequencing and bisulfite pyrosequencing showed the best all-round performance across the critical parameters of accuracy, sensitivity, and reproducibility [9]. For discovery-based research, reduced representation bisulfite sequencing (RRBS) provides a balanced approach, covering approximately 2-4 million CpGs in the human genome while being more cost-effective than whole-genome approaches [11].
For clinical applications and biomarker validation, bisulfite pyrosequencing has emerged as a preferred method due to its quantitative accuracy, reproducibility across laboratories, and compatibility with formalin-fixed paraffin-embedded (FFPE) samples [9]. The technology combines bisulfite conversion with sequencing by synthesis, providing quantitative methylation data for individual CpG sites within a targeted amplicon [31].
Experimental Protocols for Method Validation
Bisulfite Pyrosequencing Protocol
Bisulfite pyrosequencing represents one of the most robust methods for quantitative locus-specific DNA methylation analysis and serves as a gold standard for validation studies [9]. The methodology consists of three core stages:
Stage 1: DNA Treatment and Quality Control
- Extract genomic DNA using standardized protocols (e.g., DNeasy Blood and Tissue Kit)
- Assess DNA purity and concentration using spectrophotometry (A260/A280 ratio of 1.8-2.0)
- Perform bisulfite conversion using commercial kits (e.g., EZ DNA Methylation Kit from Zymo Research)
- Convert 500-1000 ng of DNA using the following program: 98°C for 10 minutes, 64°C for 2.5 hours, followed by 4°C hold [73]
- Purify converted DNA and elute in 20-30 μL of elution buffer
- Verify conversion efficiency by ensuring >99% conversion of non-CpG cytosines
Stage 2: PCR Amplification and Product Preparation
- Design primers using specialized software (MethPrimer, Bisearch) with the following parameters:
- Amplicon size: 80-200 bp
- Primer length: 20-30 bp
- Melting temperature: 60°C ± 2°C
- Include at least four non-CpG cytosines in each primer to assess bisulfite conversion
- Avoid CpG sites in primer sequences to prevent amplification bias
- Perform PCR amplification with biotinylated reverse primer
- Verify amplification success and specificity by agarose gel electrophoresis
Stage 3: Pyrosequencing and Data Analysis
- Bind biotinylated PCR products to streptavidin-coated sepharose beads
- Prepare single-stranded DNA template using the Pyrosequencing Vacuum Prep Tool
- Hybridize with sequencing primer (designed to anneal adjacent to target CpG sites)
- Perform sequencing using the Pyrosequencing instrument with nucleotide dispensation order optimized for target sequence
- Calculate methylation percentage at each CpG using Pyro Q-CpG software:
- Methylation % = C peak height / (C peak height + T peak height) × 100
- Include appropriate controls: unmethylated and in vitro methylated DNA
Diagram: Bisulfite Pyrosequencing Workflow
Reduced Representation Bisulfite Sequencing (RRBS) Protocol
RRBS provides a cost-effective approach for genome-wide DNA methylation analysis, particularly suitable for species without reference genomes and for large-scale evolutionary studies [11]. The protocol involves:
Stage 1: Library Preparation and Restriction Digestion
- Digest 5-100 ng of genomic DNA with MspI restriction enzyme (recognition site: CCGG)
- Perform size selection to enrich for 40-220 bp fragments
- Repair fragment ends and ligate methylated adapters
- Bisulfite convert DNA using optimized protocols
Stage 2: Sequencing and Bioinformatics Analysis
- Amplify libraries by PCR and sequence on Illumina platforms
- For reference-free analysis: group sequencing reads by shared restriction sites
- Calculate methylation percentages for each CpG as: mC/(mC + unmethylated C) × 100
- For cross-species comparisons: apply reference-free bioinformatic pipelines [11]
The robustness of RRBS for cross-species analysis was demonstrated in a comprehensive study of 580 animal species, which showed consistent enrichment for CpG islands across diverse taxonomic groups [11].
Advanced Applications and Emerging Technologies
Clinical Biomarker Development
DNA methylation biomarkers offer significant advantages for clinical diagnostics, including binary measurement formats, stability in stored samples, and compatibility with routine clinical workflows [9]. Successful translation requires rigorous validation through defined stages:
Analytical Validation
- Assess accuracy against a gold standard (e.g., clonal bisulfite sequencing)
- Determine sensitivity and specificity using receiver operating characteristic (ROC) analysis
- Establish limit of detection using titration series (e.g., methylated DNA spiked into unmethylated background)
- Evaluate reproducibility across operators, instruments, and laboratories [9]
Clinical Validation
- Verify performance in intended sample types (e.g., FFPE, cell-free DNA)
- Confirm tissue specificity and disease association
- Validate in independent cohorts with appropriate sample sizes
A multicenter benchmarking study demonstrated that bisulfite pyrosequencing and amplicon bisulfite sequencing consistently showed high accuracy across 32 reference samples, with the best performing assays achieving correlation coefficients >0.95 with expected values [9].
Single-Cell and Long-Read Sequencing Approaches
Emerging technologies are expanding the horizons of DNA methylation analysis:
Single-Cell DNA Methylation Analysis
- Enables characterization of epigenetic heterogeneity in complex tissues
- scRRBS (single-cell reduced representation bisulfite sequencing) provides coverage of ~1-2 million CpGs per cell [8]
- Critical for developmental biology and cancer epigenetics
Nanopore Sequencing
- Allows simultaneous detection of genetic variants and DNA methylation
- Identifies differential methylation marks in neurodevelopmental disorders [14]
- Particularly valuable for analyzing repetitive regions and structural variants
Diagram: Technology Selection Framework
Research Reagent Solutions
Selecting appropriate reagents and kits is essential for robust DNA methylation analysis. The table below outlines essential research tools for different stages of methylation analysis workflows.
Table 2: Essential Research Reagents for DNA Methylation Analysis
| Reagent/Kits | Function | Application Notes |
|---|---|---|
| DNeasy Blood & Tissue Kit (Qiagen) | DNA extraction | High-quality DNA with A260/280 >1.8; compatible with downstream bisulfite conversion [73] |
| EZ DNA Methylation Kit (Zymo Research) | Bisulfite conversion | >99% conversion efficiency; suitable for low-input samples (100 pg) [31] |
| Infinium MethylationEPIC v2.0 BeadChip (Illumina) | Genome-wide profiling | Covers >935,000 CpG sites; ideal for large cohort studies [73] |
| MethylPrimer Express Software | Primer design | Optimizes primers for bisulfite-converted DNA; critical for assay specificity [31] |
| EpiQuik DNMT Activity/Inhibition ELISA Kit | Enzyme activity assessment | High-throughput screening for drug development applications [71] |
| MethylFlash Global DNA Methylation (5-mC) ELISA Kit | Global methylation | Direct detection without DNA denaturation; species-independent [71] |
The establishment of a comprehensive validation framework for DNA methylation analysis methods reveals a sophisticated technological landscape where method selection must align with specific research objectives and required performance parameters. Locus-specific methods, particularly bisulfite pyrosequencing and amplicon bisulfite sequencing, provide the highest accuracy and reproducibility for targeted biomarker validation, while genome-wide approaches enable unbiased discovery at varying levels of genomic coverage. As the field progresses toward single-cell analyses, multi-omics integration, and clinical translation, maintaining rigorous standards for accuracy, sensitivity, reproducibility, and robustness becomes increasingly critical. By applying the structured comparison and experimental protocols outlined in this guide, researchers can make informed decisions that ensure the reliability and biological relevance of their DNA methylation data, ultimately accelerating both basic research and clinical applications in the rapidly advancing field of epigenetics.
DNA methylation analysis represents a cornerstone of epigenetic research, with methodologies broadly categorized into locus-specific and genome-wide approaches. The choice between these strategies is fundamental to experimental design, influencing everything from discovery potential to clinical applicability. Locus-specific methods provide deep, quantitative data for a predefined set of genomic regions, making them ideal for validating biomarkers or testing specific hypotheses. In contrast, genome-wide methods offer an unbiased survey of methylation patterns across the entire genome, serving as powerful discovery tools for identifying novel regulatory regions or epigenetic signatures associated with disease [8].
This guide provides a direct performance comparison of current DNA methylation analysis technologies, framing the evaluation within the critical context of selecting between targeted and comprehensive mapping strategies. We objectively compare the accuracy, coverage, cost, and throughput of leading methods, supported by recent experimental benchmarking studies. The analysis is particularly relevant for researchers and drug development professionals who must balance practical constraints like budget, sample throughput, and infrastructural capabilities against the scientific requirements of resolution, accuracy, and genomic coverage for their specific applications in basic research, biomarker development, or clinical diagnostics.
Methodologies and Experimental Protocols for DNA Methylation Assessment
The performance data presented in this guide are synthesized from multiple independent technology comparisons and benchmarking studies. The experimental protocols for generating these comparative data typically involve analyzing a common set of reference DNA samples across multiple platforms and laboratories.
For locus-specific methods, a seminal community-wide benchmarking study shipped 32 standardized reference samples to 18 participating laboratories. These samples included tumor-normal pairs, drug-treated cells, and titration series to assess sensitivity. Each laboratory independently designed assays for an average of 27 predefined genomic regions using their preferred technology—such as amplicon bisulfite sequencing (AmpliconBS), bisulfite pyrosequencing (Pyroseq), EpiTyper, or MethyLight—and returned results for centralized analysis [74].
For genome-wide approaches, comparative studies typically analyze DNA from cell lines, tissues, and whole blood using multiple platforms in parallel. A 2025 study, for instance, extracted high-quality DNA from these sources and compared four genome-wide methods: whole-genome bisulfite sequencing (WGBS), Illumina MethylationEPIC microarray, enzymatic methyl-sequencing (EM-seq), and Oxford Nanopore Technologies (ONT) sequencing. The protocols followed manufacturers' instructions with stringent quality control for DNA purity and quantity, and data were processed through standardized bioinformatic pipelines for cross-platform comparison [36] [30].
These experimental designs allow for direct comparison of technical performance metrics including accuracy, coverage, CpG detection, and concordance between methods under controlled conditions.
Comparative Performance Analysis of Major Technologies
DNA methylation analysis methods employ different biochemical principles to distinguish methylated from unmethylated cytosines. The workflow below illustrates the fundamental procedures common to bisulfite-based methods, which represent the most widely used approach.
The foundational step in most methods is bisulfite conversion, where unmethylated cytosines are deaminated to uracils while methylated cytosines remain protected. Following conversion, the DNA undergoes library preparation specific to the platform—whether for next-generation sequencing, microarray hybridization, or long-read sequencing. The final data analysis phase interprets the chemical or enzymatic signatures to determine methylation status at single-base resolution or regional levels [74] [30].
Alternative approaches are gaining traction: EM-seq uses enzymatic conversion with TET2 and APOBEC enzymes instead of harsh bisulfite chemistry, better preserving DNA integrity. Oxford Nanopore Technologies performs direct detection of DNA methylation during sequencing without prior conversion, leveraging changes in electrical current as DNA strands pass through protein nanopores [36] [30].
Quantitative Performance Comparison
The table below provides a direct comparison of key performance metrics across major DNA methylation analysis platforms, synthesizing data from multiple benchmarking studies.
| Method | Resolution | Genomic Coverage | Accuracy | Cost per Sample | Throughput | Best Applications |
|---|---|---|---|---|---|---|
| AmpliconBS | Single-base | Targeted regions | High [74] | Medium | Medium | Biomarker validation, clinical diagnostics [74] |
| Bisulfite Pyrosequencing | Single-base | Targeted regions | High [74] | Low | High | Validation studies, clinical assays [74] |
| EPIC Microarray | Single-CpG | ~850,000-935,000 CpGs | High for covered sites [36] [30] | Low | Very High | Large cohort studies, epigenome-wide association studies [36] |
| RRBS | Single-base | ~2-4 million CpGs (mammalian) [11] | High | Medium | Medium | Species without reference genomes, evolutionary studies [11] |
| WGBS | Single-base | ~80% of CpGs [36] [30] | Gold standard | High | Low | Comprehensive methylation mapping, discovery research [36] |
| EM-seq | Single-base | Comparable to WGBS | High concordance with WGBS [36] [30] | High | Low | Applications requiring high DNA integrity, uniform coverage [36] |
| Nanopore Sequencing | Single-base | Genome-wide, including repetitive regions | Moderate compared to WGBS/EM-seq [36] | Medium | Medium | Long-range methylation phasing, challenging genomic regions [36] [14] |
Key Performance Insights and Trade-offs
Accuracy and Concordance: Locus-specific methods like amplicon bisulfite sequencing and bisulfite pyrosequencing demonstrate excellent all-round performance for targeted applications, showing high accuracy in multicenter comparisons [74]. For genome-wide methods, EM-seq shows the highest concordance with the established gold standard of WGBS, while Nanopore sequencing, despite showing lower agreement, provides unique access to challenging genomic regions and enables haplotype-specific methylation analysis [36] [14].
Coverage Considerations: Microarrays like the EPIC platform provide cost-effective coverage of predefined CpG sites, particularly in regulatory regions, making them suitable for large-scale epidemiological studies [36] [30]. In contrast, sequencing-based methods offer more comprehensive coverage: RRBS efficiently captures CpG-rich regulatory regions across species, even without reference genomes [11], while WGBS and EM-seq achieve the most complete genome-wide coverage, enabling unbiased discovery of novel methylation patterns.
Practical Implementation Factors: The choice between bisulfite-based and alternative technologies involves important practical considerations. Bisulfite treatment causes substantial DNA fragmentation, potentially compromising data quality [36] [30]. EM-seq addresses this limitation through gentler enzymatic conversion, preserving DNA integrity while maintaining detection accuracy. Nanopore sequencing offers the unique advantage of simultaneous genetic and epigenetic analysis from a single run, potentially streamlining integrated analyses [14].
Essential Research Reagent Solutions
Successful DNA methylation analysis requires careful selection of reagents and kits tailored to each methodology. The table below outlines essential research solutions for major technology platforms.
| Reagent/Kits | Function | Compatible Methods |
|---|---|---|
| EZ DNA Methylation Kit (Zymo Research) | Bisulfite conversion of unmethylated cytosines | All bisulfite-based methods (AmpliconBS, Pyroseq, WGBS, RRBS) [30] |
| SQK-LSK109/LSK110 (Oxford Nanopore) | Library preparation for long-read sequencing | Nanopore sequencing for direct methylation detection [14] |
| Infinium MethylationEPIC BeadChip | Microarray-based methylation profiling | EPIC array for genome-wide methylation screening [36] [30] |
| Methylated DNA Immunoprecipitation (MeDIP) Kits | Antibody-based enrichment of methylated DNA | MeDIP-seq for methylome profiling [51] |
| NEBN EM-Seq Kit | Enzymatic conversion of unmodified cytosines | EM-seq for bisulfite-free methylation detection [36] |
| Methylation-Specific PCR Assays | Targeted amplification of methylated sequences | locus-specific validation (qMSP, MethyLight) [74] |
Strategic Guidance for Method Selection
Decision Framework for Technology Selection
Choosing the appropriate DNA methylation analysis method requires careful consideration of research goals, sample characteristics, and practical constraints. The decision workflow below outlines a systematic approach for method selection.
Application-Optimized Recommendations
Large Cohort Epigenome-Wide Association Studies: For studies involving thousands of samples where budget constraints are significant but predefined genomic coverage is sufficient, the Infinium EPIC microarray offers the optimal balance of cost, throughput, and coverage [36] [30].
Comprehensive Methylome Discovery Research: For discovery-oriented projects requiring base-resolution, genome-wide data, WGBS remains the gold standard, though EM-seq is emerging as a superior alternative due to better DNA preservation and more uniform coverage [36].
Evolutionary Studies and Cross-Species Analysis: For comparative methylomics across species, particularly those without reference genomes, RRBS provides a robust solution that efficiently captures conserved regulatory regions while being cost-effective for multiple samples [11].
Clinical Biomarker Validation and Diagnostics: For translating methylation biomarkers into clinical applications, bisulfite pyrosequencing and amplicon bisulfite sequencing offer the required accuracy, reproducibility, and quantitative precision for routine use [74].
Integrated Genetic-Epigenetic Analysis: For studies requiring simultaneous assessment of genetic variants and methylation patterns, Nanopore sequencing enables both analyses in a single run, providing haplotype-specific methylation data [14].
The evolving landscape of DNA methylation technologies offers researchers multiple pathways for epigenetic investigation, each with distinct strengths and limitations. Locus-specific methods provide the precision, sensitivity, and throughput required for clinical applications and focused validation studies, while genome-wide approaches deliver the comprehensive coverage essential for discovery science. Emerging technologies like EM-seq and nanopore sequencing address limitations of traditional bisulfite-based methods, particularly regarding DNA degradation and limited sequence context.
The optimal choice depends fundamentally on the research question, with practical considerations including sample number, DNA quality and quantity, bioinformatic capabilities, and budget constraints. As the field advances toward more integrated analyses of multiple epigenetic layers and single-cell resolution, we can anticipate continued methodological refinements that further enhance accuracy, reduce costs, and expand our ability to decipher the complex language of DNA methylation in health and disease.
DNA methylation, the addition of a methyl group to cytosine bases, is a fundamental epigenetic mechanism regulating gene expression and genome integrity. Its analysis relies on two primary methodological philosophies: locus-specific and genome-wide approaches. Locus-specific techniques focus on quantifying DNA methylation at predefined genomic regions, often with high sensitivity and cost-effectiveness, making them suitable for clinical assay development and biomarker validation [9]. In contrast, genome-wide methods provide an unbiased survey of methylation patterns across the entire genome, enabling novel discovery and comprehensive epigenomic profiling [8] [11]. The choice between these strategies carries significant implications for diagnostic accuracy, clinical utility, and research outcomes in fields such as oncology, prenatal medicine, and pharmacology. This guide objectively compares the performance of these methodologies through experimental data and detailed case studies, providing a framework for researchers and clinicians to select the optimal approach for their specific applications.
Methodological Comparison and Benchmarking Data
A community-wide benchmarking study systematically evaluated the performance of DNA methylation assays compatible with routine clinical use [9]. The study involved 18 laboratories analyzing 32 reference samples, collectively contributing 21 locus-specific assays and six global assays. The evaluation assessed sensitivity on low-input samples and the ability to discriminate between cell types.
Table 1: Performance Characteristics of Major DNA Methylation Analysis Technologies
| Technology | Analysis Type | Throughput | DNA Input | Best Applications | Key Advantages | Key Limitations |
|---|---|---|---|---|---|---|
| Amplicon Bisulfite Sequencing [9] | Locus-specific | High | Low | Biomarker validation, clinical diagnostics | Excellent all-round performance, single-CpG resolution | Limited to targeted regions |
| Bisulfite Pyrosequencing [9] | Locus-specific | Moderate | Low | Clinical assays, biomarker validation | Quantitative accuracy, rapid turnaround | Limited multiplexing capability |
| Methylation-Specific PCR [9] | Locus-specific | High | Very Low | High-throughput clinical screening | Extreme sensitivity for rare methylation events | Semi-quantitative, design challenges |
| Whole-Genome Bisulfite Sequencing [49] | Genome-wide | Highest | High (≥100 ng) | Discovery research, comprehensive profiling | Single-base resolution, entire methylome coverage | High cost, complex data analysis |
| Reduced Representation Bisulfite Sequencing [11] [49] | Genome-wide | Moderate | Moderate (≥30 ng) | Epigenome-wide association studies | Cost-effective for CpG-rich regions | Covers only ~10% of CpGs |
| Methylation Microarrays [49] | Genome-wide | High | Moderate (≥250 ng) | Large-scale cohort studies | Excellent for population-level studies | Limited to predefined CpG sites |
The benchmarking revealed that amplicon bisulfite sequencing and bisulfite pyrosequencing demonstrated the best all-round performance for locus-specific applications, offering an optimal balance of accuracy, sensitivity, and practical implementation [9]. For discovery-phase research, genome-wide methods like whole-genome bisulfite sequencing and reduced representation bisulfite sequencing provided the most comprehensive data, albeit with higher resource requirements.
Table 2: Quantitative Performance Metrics from Benchmarking Studies
| Method | Accuracy (Correlation with Reference) | Sensitivity | Reproducibility (CV) | Cost per Sample | Hands-on Time |
|---|---|---|---|---|---|
| AmpliconBS | >0.95 | High (detects ≥1% methylation) | <5% | $$ | Moderate |
| Bisulfite Pyrosequencing | >0.98 | High (detects ≥5% methylation) | <3% | $ | Low |
| Methylation-Specific PCR | 0.85-0.95 | Very High (detects ≥0.1% methylation) | 5-10% | $ | Low |
| RRBS | >0.90 | Moderate | 5-8% | $$$ | High |
| Infinium MethylationEPIC | >0.95 | Moderate | <5% | $$ | Low |
The data demonstrates a fundamental trade-off: locus-specific methods excel in sensitivity, cost-effectiveness, and clinical implementation, while genome-wide approaches provide unbiased discovery power and comprehensive profiling. The optimal choice depends fundamentally on the research or clinical question, with locus-specific methods being preferable for validated biomarker panels and genome-wide methods being essential for novel discovery.
Experimental Protocols for DNA Methylation Analysis
Core Technical Workflows
The following diagram illustrates the fundamental experimental workflows for major DNA methylation analysis technologies:
Detailed Methodological Protocols
High-Resolution Methyl-Capture Sequencing (MC-seq) Protocol
The MC-seq protocol for genome-wide methylation profiling, as applied in trisomy 18 research [75], involves:
Library Preparation and Target Capture:
- DNA Fragmentation: 3μg of genomic DNA is fragmented using adaptive focused acoustic (AFA) technology.
- Library Construction: Fragmented DNA undergoes end-repair, A-tailing, and ligation of Agilent adapters followed by PCR amplification.
- Target Capture: 250ng of library is hybridized with SureSelect Human Methyl-Seq capture probes at 65°C for 24 hours.
- Bisulfite Conversion: Captured DNA is treated with Zymo EZ DNA Methylation-Gold Kit for cytosine conversion.
- Final Amplification: 8 cycles of PCR enrich adapter-ligated fragments, plus 6 additional cycles for barcode incorporation.
Data Analysis:
- Quality Control: FastQC (v0.11.5) assesses read quality, followed by adapter trimming with Trimmomatic.
- Alignment: Cleaned reads are aligned to reference genome (hg19) using BSMAP with parameters -n 1 and -r 0.
- Methylation Calling: Methylation ratios are calculated with methratio.py (BSMAP) for each cytosine with ≥10x coverage.
- DMR Identification: Differentially methylated regions are identified using criteria of |Δmean| ≥ 0.2, P-value < 0.05, and FDR ≤ 0.05.
SERS-Based Methylation Detection Protocol
For label-free methylation detection in cancer diagnostics [76]:
Sample Preparation:
- DNA Extraction: Genomic DNA is purified using kits such as PureLink Genomic DNA Mini Kit, with Proteinase K and RNase A treatment.
- Cation Optimization: DNA is mixed with 5×10⁻⁴ M Ca²⁺ to enhance adsorption to silver nanoparticles.
- Substrate Preparation: Silver nanoparticles are synthesized and characterized for optimal plasmonic properties.
SERS Measurement:
- Spectral Acquisition: Raman spectra are collected with 785nm laser excitation, 10s integration time.
- Methylation Quantification: The intensity ratio of 790 cm⁻¹ band (5-methylcytosine marker) to 1090 cm⁻¹ band (DNA backbone) is calculated.
- Validation: Results are correlated with bisulfite sequencing data to establish calibration curves (R = 0.94, p = 0.005).
Cancer Diagnostics: Early Detection via Liquid Biopsy
Clinical Application and Experimental Findings
In cancer diagnostics, DNA methylation analysis of circulating tumor DNA (ctDNA) enables non-invasive liquid biopsy for early detection, monitoring, and personalized treatment. Breast cancer research demonstrates how methylation changes in tumor suppressor genes (e.g., BRCA1, RASSF1A) occur early in carcinogenesis, preceding genetic alterations [49].
A 2025 review highlighted advancements in detecting tumor-specific ctDNA methylation in blood, with distinct signatures differentiating breast cancer subtypes like triple-negative breast cancer (TNBC) [49]. However, detecting low-abundance ctDNA in early-stage cancers requires extremely sensitive methods, favoring targeted locus-specific approaches for clinical application.
Research comparing methylation detection technologies found bisulfite pyrosequencing and amplicon bisulfite sequencing superior for clinical diagnostics, demonstrating high accuracy and robustness in multicenter studies [9]. Emerging techniques like SERS-based detection offer promising alternatives, identifying the 790 cm⁻¹ band as a specific marker for 5-methylcytosine with strong correlation to methylation levels (R=0.94, p=0.005) [76].
Machine Learning for Cross-Tissue Methylation Prediction
A 2024 study developed machine learning models to predict locus-specific DNA methylation in cancerous tissues (CT) using paracancerous tissues (PT) as surrogates [77]. The XGBoost algorithm achieved significant improvement in mean absolute error at >68% of CpG sites, enhancing statistical power in differential methylation analysis. This approach is particularly valuable when tumor tissue is difficult to obtain through biopsy, addressing clinical sampling challenges in prostate, lung, and cholangiocarcinoma malignancies [77].
Prenatal Testing: Non-Invasive Detection of Chromosomal Abnormalities
Experimental Design and Methylation Findings
Non-invasive prenatal testing (NIPT) has been transformed by DNA methylation analysis of cell-free fetal DNA. A 2025 study investigated trisomy 18 (T18)-specific methylation patterns in first-trimester chorionic villi, revealing distinct epigenetic signatures [75].
Experimental Approach:
- Sample Collection: Chorionic villi from 5 T18 and 5 euploid fetuses at 11-13 weeks gestation.
- Methylation Profiling: High-resolution methyl-capture sequencing (MC-seq) assessing ~3.2 million CpG sites.
- Data Analysis: Identification of differentially methylated CpGs (DMCs) and regions (DMRs) with functional enrichment analysis.
Key Findings:
- Global Hypermethylation: T18 samples showed widespread hypermethylation versus controls.
- 6,510 DMCs Identified: 4,022 hypermethylated and 2,488 hypomethylated sites.
- Chromosome 18 Enrichment: The trisomic chromosome contained the highest number of hypermethylated DMRs.
- 76 Biomarker Candidates: DMRs with inverse methylation patterns in maternal blood were identified as potential NIPT targets.
The study demonstrated that methylation alterations represent downstream consequences of chromosomal imbalance and provide measurable signals for non-invasive detection [75]. The locus-specific nature of these biomarkers makes them ideally suited for targeted clinical assay development.
Technology Selection for Prenatal Applications
In prenatal diagnostics, locus-specific methods like methylation-specific PCR and targeted bisulfite sequencing dominate clinical applications due to their:
- High sensitivity for detecting fetal DNA in maternal circulation
- Cost-effectiveness for population screening
- Rapid turnaround time essential for clinical decision-making
- Ability to focus on validated biomarker panels
The research-to-clinical translation pipeline typically begins with genome-wide discovery (MC-seq or RRBS) to identify candidate biomarkers, followed by development of targeted locus-specific assays for clinical implementation [75].
Drug Development: Predicting Therapeutic Response
Pharmacoepigenetics and Adverse Drug Response
DNA methylation analysis plays an increasingly important role in pharmacoepigenetics, helping to explain inter-individual variation in drug response. A 2025 study identified DNA methylation sites affecting responses to anticoagulant and cardiometabolic drugs using electronic health record-derived GWAS summary statistics integrated with methylation quantitative trait loci (mQTL) data [78].
Key Findings:
- Warfarin: 71 DNA methylation sites identified, with 8 near VKORC1 and 48 on chromosome 6 near HLA genes.
- Statins: 17 methylation sites identified, 8 near APOB gene (involved in LDL cholesterol metabolism).
- Methodology: Instrumental variable analysis integrated UK Biobank pharmacogenomic GWAS with GoDMC mQTL data.
This approach demonstrates how epigenome-wide association studies can identify methylation biomarkers predictive of drug response, potentially minimizing adverse drug reactions (ADRs) that cause ~7,000 deaths annually [78].
Multi-Omics Integration in Drug Discovery
Network-based multi-omics integration methods have revolutionized drug discovery by combining DNA methylation data with other molecular layers (genomics, transcriptomics, proteomics) [79]. These approaches can be categorized into:
- Network Propagation/Diffusion Models
- Similarity-Based Approaches
- Graph Neural Networks
- Network Inference Models
The integration of DNA methylation data within biological networks (protein-protein interactions, metabolic pathways) enables better prediction of drug targets, therapeutic response, and drug repurposing opportunities [79]. DNA methylation provides particularly valuable information for understanding drug resistance mechanisms and cellular heterogeneity in treatment response.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Solutions for DNA Methylation Analysis
| Reagent/Solution | Function | Example Products | Application Notes |
|---|---|---|---|
| Bisulfite Conversion Kits | Chemical conversion of unmethylated cytosines to uracils | Zymo EZ DNA Methylation-Gold Kit | Critical step for most methods; optimized protocols vary by input DNA quality and quantity |
| Methylation-Specific Restriction Enzymes | Differential digestion based on methylation status | Mspl, HpaII | Used in RRBS and locus-specific methods; sensitivity to CpG context important |
| Capture Probes for Targeted Sequencing | Enrichment of specific genomic regions | Agilent SureSelect Methyl-Seq | Enable focused methylation profiling; design considerations for CpG coverage |
| 5-methylcytosine Antibodies | Immunoprecipitation of methylated DNA | Diagenode Anti-5-mC Antibody | Used in MeDIP-seq; bias toward low-CpG density regions |
| Methylated DNA Standards | Quality control and quantification | Millipore Sigma Methylated DNA | Essential for assay validation and cross-platform comparisons |
| Methylation-Sensitive PCR Reagents | Amplification of methylation-specific targets | Qiagen MSP Kit | Optimized for discrimination of methylated/unmethylated templates |
| SERS Nanoparticles | Signal enhancement for label-free detection | Silver nanoparticles with Ca²⁺ activation | Cation-mediated adsorption critical for DNA detection [76] |
| Whole-Genome Amplification Kits | Amplification of limited DNA samples | REPLI-g Advanced DNA Kit | Important for low-input applications; potential methylation bias |
Integrated Workflow for Method Selection
The following diagram illustrates a decision framework for selecting appropriate DNA methylation analysis methods based on research goals and practical constraints:
The comparative analysis of DNA methylation methodologies across cancer diagnostics, prenatal testing, and drug development reveals a consistent pattern: genome-wide approaches drive discovery while locus-specific methods enable clinical implementation. The optimal methodology depends on the specific application phase, with research-oriented questions benefiting from comprehensive epigenomic profiling and clinical applications requiring targeted, robust, and cost-effective assays.
Emerging technologies like bisulfite-free sequencing (EM-seq, TAPS) and direct detection methods (SERS, nanopore sequencing) promise to overcome limitations of conventional approaches, particularly for analyzing degraded or low-input samples [49] [76]. Furthermore, machine learning integration and multi-omics network analysis are enhancing our ability to extract biological insights from DNA methylation data, particularly in complex applications like drug response prediction [79] [77].
As the field advances, the convergence of methodological improvements in both locus-specific and genome-wide technologies will continue to expand applications in clinical diagnostics and therapeutic development, ultimately enabling more precise and personalized medical interventions based on epigenetic profiling.
Selecting the appropriate analytical method is a critical step in research that directly impacts the validity, cost, and ultimate success of a study. In the field of DNA methylation analysis, this decision is particularly complex, with researchers often facing a fundamental choice between locus-specific and genome-wide approaches. Each method offers distinct advantages and limitations based on the research objectives, resources, and experimental constraints.
A decision matrix (also known as a prioritization matrix or weighted scoring model) provides a structured framework for evaluating and prioritizing a list of options based on multiple, weighted criteria [80]. This qualitative technique helps rank multi-dimensional options, minimizing subjective bias and bringing transparency to the decision-making process [81]. In this guide, we will demonstrate how to apply a decision matrix to select the most appropriate DNA methylation analysis method for your research goals, using the comparison of locus-specific versus genome-wide approaches as our case study.
Understanding DNA Methylation Analysis Methods
DNA methylation—the addition of a methyl group to cytosine bases—is a fundamental epigenetic modification influencing gene expression, cellular differentiation, and disease pathogenesis [8] [11]. The two primary methodological approaches for its investigation serve complementary but distinct research purposes.
Genome-wide DNA methylation analysis provides an unbiased, comprehensive profile of methylation patterns across the entire genome. Techniques include:
- Whole Genome Bisulfite Sequencing (WGBS): Provides single-base resolution methylation measurements across the entire genome [8] [11].
- Reduced Representation Bisulfite Sequencing (RRBS): Offers a cost-effective alternative by enriching for CpG-rich regions, covering approximately 4 million of the 28 million CpGs in the human genome [11].
- Methylation arrays (e.g., Illumina EPIC arrays): Interrogate pre-defined sets of CpG sites (over 935,000 sites for EPIC v2.0) using hybridization-based technology [73].
Locus-specific DNA methylation analysis focuses on predetermined genomic regions of interest with typically higher depth and lower cost per region. Techniques include:
- Bisulfite sequencing PCR (BSP): Traditional method for targeted sequencing of specific regions after bisulfite conversion [8].
- Nanopore adaptive targeted long-read sequencing: Enables real-time enrichment and simultaneous assessment of genetic and epigenetic information at specific loci [14].
- Methylation-specific PCR (MSP): Rapid detection of methylation status at specific CpG sites.
Table: Comparison of DNA Methylation Analysis Methods
| Feature | Genome-Wide Approaches | Locus-Specific Approaches |
|---|---|---|
| Genomic Coverage | Comprehensive (entire genome or enriched regions) | Focused (pre-defined regions only) |
| Resolution | Single-base (sequencing) or probe-based (arrays) | Typically single-base resolution |
| Cost per sample | Higher ($ hundreds to thousands) | Lower ($ tens to hundreds) |
| Sample throughput | Moderate to high (especially arrays) | Low to moderate |
| Data complexity | High, requires advanced bioinformatics | Moderate, more accessible analysis |
| Ideal application | Discovery phase, biomarker identification, epigenome-wide association studies | Validation studies, diagnostic applications, focused hypothesis testing |
| Required DNA input | Varies (50-1000ng) | Typically lower (10-100ng) |
The Decision Matrix: A Framework for Method Selection
What is a Decision Matrix?
A decision matrix is a systematic tool that helps evaluate and prioritize multiple alternatives against a set of weighted criteria [80]. Also known as the Pugh method or weighted scoring model, it brings objectivity and transparency to complex decision-making processes [81]. In research method selection, it helps quantify subjective judgments, minimizes personal biases, and enables clear comparison of competing approaches.
Step-by-Step Guide to Building Your Decision Matrix
Step 1: Define Your Decision Context
Clearly articulate the specific research problem. For our example: "Which DNA methylation analysis method should I select for my research on identifying differential methylation in a specific gene pathway in cancer versus normal tissues?"
Step 2: List Your Alternatives
Identify the potential methods to evaluate. In our case:
- Alternative A: Genome-wide approach (RRBS)
- Alternative B: Locus-specific approach (Nanopore adaptive T-LRS)
Step 3: Determine Evaluation Criteria
Establish the factors that will influence your decision. For methylation method selection, consider:
- Analytical resolution and specificity
- Genome coverage relevant to your hypothesis
- Cost efficiency and budget constraints
- Technical feasibility and expertise required
- Sample throughput requirements
- Compatibility with your sample type and quality
- Data analysis complexity and bioinformatic requirements
Step 4: Assign Weights to Criteria
Rank criteria by importance, assigning weights that total 100%. The more important the criterion, the higher its weighting [81].
Table: Example Criteria Weighting for DNA Methylation Method Selection
| Criterion | Weight | Rationale for Weighting |
|---|---|---|
| Analytical resolution | 25% | Critical for detecting biologically relevant methylation changes |
| Cost efficiency | 20% | Must align with available resources; affects sample size |
| Technical feasibility | 15% | Must be implementable with available expertise and equipment |
| Hypothesis relevance | 15% | Must adequately address the research question |
| Sample compatibility | 10% | Must work with available sample type and quality |
| Data complexity | 10% | Must align with available bioinformatics capabilities |
| Throughput | 5% | Must complete analysis within required timeframe |
Step 5: Score Each Alternative
Evaluate each method against all criteria using a consistent scoring system (e.g., 1-5 scale, where 5 = excellent).
Table: Example Scoring of DNA Methylation Methods
| Criterion | Weight | Genome-Wide (RRBS) | Locus-Specific (Nanopore) |
|---|---|---|---|
| Analytical resolution | 25% | 4 (Covers millions of CpGs at single-base resolution) [11] | 5 (Single-base resolution with haplotype information) [14] |
| Cost efficiency | 20% | 2 (Higher cost per sample) | 4 (More cost-effective for targeted regions) |
| Technical feasibility | 15% | 3 (Requires specialized expertise) | 4 (Newer but increasingly accessible) |
| Hypothesis relevance | 15% | 3 (May generate excess irrelevant data) | 5 (Directly targets regions of interest) |
| Sample compatibility | 10% | 4 (Works with various sample types) | 3 (May have specific DNA quality requirements) |
| Data complexity | 10% | 2 (Requires advanced bioinformatics) | 3 (Moderate analysis requirements) |
| Throughput | 5% | 4 (Moderate to high throughput) | 3 (Moderate throughput) |
Step 6: Calculate Weighted Scores and Analyze Results
Multiply each score by its criterion weight and sum for each alternative.
Table: Weighted Score Calculation
| Criterion | Weight | Genome-Wide Score | Weighted | Locus-Specific Score | Weighted |
|---|---|---|---|---|---|
| Analytical resolution | 25% | 4 | 1.00 | 5 | 1.25 |
| Cost efficiency | 20% | 2 | 0.40 | 4 | 0.80 |
| Technical feasibility | 15% | 3 | 0.45 | 4 | 0.60 |
| Hypothesis relevance | 15% | 3 | 0.45 | 5 | 0.75 |
| Sample compatibility | 10% | 4 | 0.40 | 3 | 0.30 |
| Data complexity | 10% | 2 | 0.20 | 3 | 0.30 |
| Throughput | 5% | 4 | 0.20 | 3 | 0.15 |
| TOTAL | 100% | 3.10 | 4.15 |
Based on this analysis, the locus-specific approach emerges as the preferred method for this specific research scenario, with a total weighted score of 4.15 versus 3.10 for the genome-wide approach.
Experimental Protocols for Method Validation
Once a method is selected, proper validation is essential. The comparison of methods experiment estimates inaccuracy or systematic error by analyzing patient samples by both new and comparative methods [82].
Protocol for Method Comparison Studies
Sample Selection and Preparation:
- Select a minimum of 40 patient specimens covering the entire working range of the method [82]
- Include the spectrum of diseases expected in routine application
- Analyze specimens within two hours of each other by test and comparative methods to ensure stability [82]
- For methylation studies, use consistent DNA extraction protocols (e.g., DNeasy Blood and Tissue Kit) [73] and quality control measures (e.g., Nanodrop spectrophotometry)
Experimental Design:
- Perform measurements over multiple days (minimum of 5 recommended) to minimize run-to-run variability [82]
- Consider duplicate measurements to identify sample mix-ups, transposition errors, and other mistakes
- For methylation analysis, include both positive and negative controls in each run
Data Analysis:
- Graph data using difference plots (test minus comparative results vs. comparative result) for visual inspection [82]
- Calculate linear regression statistics (slope, y-intercept, standard deviation about the line) for wide analytical ranges
- For narrow analytical ranges, calculate average difference (bias) between methods
- Determine systematic error at medically or biologically relevant decision concentrations
Decision Workflow Visualization
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Research Reagent Solutions for DNA Methylation Analysis
| Reagent/Kit | Function | Application Notes |
|---|---|---|
| DNeasy Blood & Tissue Kit (Qiagen) | High-quality DNA extraction from various sample types | Essential for obtaining high-molecular-weight DNA; critical for long-read sequencing [14] |
| EZ DNA Methylation Kit (Zymo Research) | Bisulfite conversion of unmethylated cytosines | Gold standard for pre-sequencing conversion; efficiency impacts data quality [73] |
| SQK-LSK109/LSK110 Kit (Oxford Nanopore) | Library preparation for nanopore sequencing | Enables detection of base modifications including 5-methylcytosine during sequencing [14] |
| SMRTbell Prep Kit 3.0 (PacBio) | Library preparation for HiFi sequencing | Provides long reads for comprehensive methylation analysis in complex regions |
| Methylation EPIC v2.0 BeadChip (Illumina) | Genome-wide methylation profiling using arrays | Interrogates >935,000 CpG sites; cost-effective for large cohort studies [73] |
| Short-read Eliminator Kit (PacBio) | Depletion of short DNA fragments | Enhances sequencing of long DNA fragments for improved genome assembly [14] |
| ModKit software | Analysis of modified base data from nanopore sequencing | Quantifies 5-methylcytosine from nanopore data; generates bedMethyl format tables [14] |
| ChAMP R Package | Preprocessing and analysis of methylation array data | Implements BMIQ normalization; handles batch effect correction and quality control [73] |
The decision matrix provides a robust, transparent framework for selecting the most appropriate DNA methylation analysis method for your specific research goals. By systematically evaluating genome-wide and locus-specific approaches against weighted criteria relevant to your research context, you can make an objective, defensible choice that optimizes resources and maximizes the likelihood of scientific success.
As demonstrated in our case study, locus-specific methods like nanopore adaptive targeted sequencing may be preferable for focused hypothesis testing, while genome-wide approaches remain invaluable for discovery-phase research. The optimal choice ultimately depends on your specific research question, resources, and experimental constraints. By applying this structured decision-making approach, researchers can navigate the complex landscape of epigenetic分析方法选择with greater confidence and scientific rigor.
Conclusion
The choice between locus-specific and genome-wide DNA methylation analysis is not a matter of one being superior to the other, but rather a strategic decision dictated by the research question. Locus-specific methods like pyrosequencing offer cost-effective, high-throughput validation for defined targets, while genome-wide approaches like WGBS and EM-seq are indispensable for unbiased discovery. Emerging long-read sequencing technologies promise to bridge this divide by offering both single-base resolution and long-range epigenetic information. As we move forward, the integration of methylation data with other omics layers and its application in single-cell analysis and liquid biopsies will further solidify DNA methylation's role in precision medicine, driving advancements in diagnostic biomarkers and therapeutic development.
References
- 1. - 5 - Wikipedia Methylcytosine [https://en.wikipedia.org/wiki/5-Methylcytosine]
- 2. Frontiers | Species-Specific 5 mC and 5 hmC Genomic Landscapes ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2018.00039/full]
- 3. Current and Emerging Technologies for the Analysis of the... [https://pubmed.ncbi.nlm.nih.gov/27826846/]
- 4. Quantitative comparison of DNA ... | Nature Biotechnology methylation [https://www.nature.com/articles/nbt.3605?error=cookies_not_supported&code=ecb8d6fe-9095-4724-9efe-2fad79b884ca]
- 5. Frontiers | DNA methylation biomarker analysis from low-survival-rate cancers based on genetic functional approaches [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1523524/full]
- 6. A comprehensive long-read sequencing system to assess DNA methylation at differentially methylated regions and imprinting-disorder-related genes | Genome Medicine | Full Text [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-025-01559-w]
- 7. Advances in RNA cytosine- methylation: detection, regulatory... [https://link.springer.com/article/10.1186/s40364-020-00225-0]
- 8. Current and Emerging Technologies for the Analysis of the... [https://pubmed.ncbi.nlm.nih.gov/36350519/]
- 9. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications | Nature Biotechnology [https://www.nature.com/articles/nbt.3605]
- 10. Profiling genome-wide DNA methylation | Epigenetics & Chromatin | Full Text [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-016-0075-3]
- 11. analysis of Comparative -scale, base-resolution genome ... DNA [https://www.nature.com/articles/s41467-022-34828-y?error=cookies_not_supported&code=b70a5837-c753-46e7-83a8-7c2a59833490]
- 12. DNA Methyltransferase Inhibitors and Their Emerging Role in Epigenetic Therapy of Cancer | Anticancer Research [https://ar.iiarjournals.org/content/33/8/2989]
- 13. Leveraging locus - specific epigenetic heterogeneity to improve the... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7574234/]
- 14. Diagnostic utility of single- locus mark in Sotos... DNA methylation [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-025-01832-0]
- 15. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9067529/]
- 16. Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons - PubMed [https://pubmed.ncbi.nlm.nih.gov/33975521/]
- 17. Advancements in DNA technologies and their application... methylation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12309559/]
- 18. A Comparative Overview of Epigenomic Profiling Methods - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8340004/]
- 19. The technology landscape for detection of DNA methylation in cancer liquid biopsies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11792835/]
- 20. Comparative performance evaluation of bisulfite - and enzyme-based... [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-025-01855-7]
- 21. Sequencing (BS-Seq)/WGBS Bisulfite [https://emea.illumina.com/techniques/sequencing/methylation-sequencing/bisulfite-sequencing.html]
- 22. Affinity-Based Enrichment Techniques for the Genome-Wide Analysis of 5-Hydroxymethylcytosine - PubMed [https://pubmed.ncbi.nlm.nih.gov/29224170/]
- 23. Affinity-based enrichment strategies to assay methyl-CpG binding activity and DNA methylation in early Xenopus embryos - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3169474/]
- 24. DNA Methylation Analysis: Choosing the Right Method - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4810160/]
- 25. A Practical Guide to the Measurement and Analysis of DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6775954/]
- 26. Sequencing | NGS advantages Methylation [https://www.illumina.com/techniques/sequencing/methylation-sequencing.html]
- 27. Understanding DNA Sequencing... | Celemics, Inc. Methylation [https://www.celemics.com/resources/blogs/understanding-dna-methylation-sequencing/]
- 28. DNA Methylation Analysis | Genome-wide methylation profiling [https://www.illumina.com/techniques/multiomics/epigenetics/dna-methylation-analysis.html]
- 29. DNA methylation data by sequencing: experimental approaches and recommendations for tools and pipelines for data analysis | Clinical Epigenetics | Full Text [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-019-0795-x]
- 30. Comparison of current methods for genome-wide DNA methylation profiling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12376410/]
- 31. DNA Methylation Validation Methods: a Coherent Review with Practical... [https://biologicalproceduresonline.biomedcentral.com/articles/10.1186/s12575-019-0107-z]
- 32. of Comparison and Methylation-Specific... Bisulfite Pyrosequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC7730915/]
- 33. Study of Comparison - MS and HRM ... | PLOS One Pyrosequencing [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0052501]
- 34. Assessing combined methylation-sensitive high resolution melting and... [https://pubmed.ncbi.nlm.nih.gov/21364322/]
- 35. Characterisation and reproducibility of the HumanMethylationEPIC... [https://pubmed.ncbi.nlm.nih.gov/38448820/]
- 36. of current methods for Comparison -wide DNA genome ... methylation [https://pubmed.ncbi.nlm.nih.gov/40855329/]
- 37. of current methods for genome-wide DNA Comparison ... methylation [https://link.springer.com/article/10.1186/s13072-025-00616-3]
- 38. Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing | Scientific Reports [https://www.nature.com/articles/s41598-019-46875-5]
- 39. (BS-Seq)/ Bisulfite Sequencing WGBS [https://www.illumina.com/techniques/sequencing/methylation-sequencing/bisulfite-sequencing.html]
- 40. A comparison of DNA methylation detection between HiFi... [https://pubmed.ncbi.nlm.nih.gov/40763129/]
- 41. sequencing detects Enzymatic at... methyl DNA methylation [https://pmc.ncbi.nlm.nih.gov/articles/PMC8256858/]
- 42. Practical Case Studies for Comparison Between EM - seq and Other... [https://www.cd-genomics.com/resource-em-seq-comparison-wgbs-pbat-case.html]
- 43. A comparison of methods for detecting DNA methylation from long-read sequencing of human genomes | Genome Biology | Full Text [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03207-9]
- 44. of current methods for genome-wide Comparison ... DNA methylation [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-025-00616-3]
- 45. DNA 5-methylcytosine detection and methylation phasing using PacBio circular consensus sequencing | Nature Communications [https://www.nature.com/articles/s41467-023-39784-9]
- 46. Comparative analysis of genome-scale, base-resolution DNA methylation profiles across 580 animal species | Nature Communications [https://www.nature.com/articles/s41467-022-34828-y]
- 47. Estimating genome-wide DNA methylation heterogeneity with methylation patterns | Epigenetics & Chromatin | Full Text [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-023-00521-7]
- 48. From concept to clinic: a roadmap for DNA methylation biomarkers in liquid biopsies | Oncogene [https://www.nature.com/articles/s41388-025-03624-5]
- 49. in breast cancer: early detection and DNA ... methylation biomarker [https://pmc.ncbi.nlm.nih.gov/articles/PMC12020129/]
- 50. in breast cancer: early detection and DNA ... methylation biomarker [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06495-2]
- 51. Genome-wide mapping of DNA methylation: a quantitative technology comparison - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3066564/]
- 52. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications - PubMed [https://pubmed.ncbi.nlm.nih.gov/27347756/]
- 53. Quantitative comparison of DNA assays for methylation ... biomarker [https://www.nature.com/articles/nbt.3605?error=cookies_not_supported]
- 54. Integrated analysis of genome - wide ... | Oncotarget DNA methylation [https://www.oncotarget.com/article/11534/text/]
- 55. Leveraging locus - specific epigenetic heterogeneity to improve the... [https://link.springer.com/article/10.1186/s13148-020-00939-w]
- 56. Current and Emerging Technologies for the Analysis of... | SpringerLink [https://link.springer.com/chapter/10.1007/978-3-319-43624-1_15]
- 57. Interpretation of genome - wide infinium methylation data from ligated... [https://bmcresnotes.biomedcentral.com/articles/10.1186/1756-0500-5-117]
- 58. A methodological study of genome - wide analyses... DNA methylation [https://www.oncotarget.com/article/14739/text/]
- 59. Genome-wide cell-free DNA methylation analyses improve accuracy of non-invasive diagnostic imaging for early-stage breast cancer | Molecular Cancer | Full Text [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-021-01330-w]
- 60. Epigenetics-targeted drugs: current paradigms and future challenges | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-024-02039-0]
- 61. Comparative performance evaluation of bisulfite - and enzyme-based... [https://pubmed.ncbi.nlm.nih.gov/40181442/]
- 62. Ultra-mild bisulfite outperforms existing... | Nature Communications [https://www.nature.com/articles/s41467-025-66033-y?error=cookies_not_supported&code=2f631cc4-6938-4ad1-ba09-95b54c203892]
- 63. An optimized rapid bisulfite method with high recovery of... conversion [https://bmcmolbiol.biomedcentral.com/articles/10.1186/s12867-017-0101-4]
- 64. Methods for analysis of specific DNA methylation status - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1046202320300979]
- 65. Principles and challenges of genome-wide DNA methylation analysis | Nature Reviews Genetics [https://www.nature.com/articles/nrg2732]
- 66. Systematic evaluation of cell-type deconvolution pipelines for... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9294431/]
- 67. Impact of DNA methylation on 3D genome structure | Nature Communications [https://www.nature.com/articles/s41467-021-23142-8]
- 68. A DNA methylation database of human and mouse hematological malignancy cell lines | Leukemia [https://www.nature.com/articles/s41375-024-02478-2]
- 69. Research DNA - Methylation of Advantages... Methods Comparison [https://www.linkedin.com/pulse/dna-methylation-research-methods-comparison-advantages-disadvantages-aduoc]
- 70. DNA Methylation-Targeted Drugs - PubMed [https://pubmed.ncbi.nlm.nih.gov/28926427/]
- 71. DNA Methylation and Drug Discovery: Unveiling Epigenetic Mechanisms for Therapeutic Breakthroughs | EpigenTek [https://www.epigentek.com/catalog/dna-methylation-and-drug-discovery-unveiling-epigenetic-mechanisms-for-therapeutic-breakthroughs-n-93.html]
- 72. Methods in DNA methylation array dataset analysis: A review - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11153885/]
- 73. Exploration of Epigenetic Mechanisms and Biomarkers among Patients... [https://www.dovepress.com/exploration-of-epigenetic-mechanisms-and-biomarkers-among-patients-wit-peer-reviewed-fulltext-article-NDT]
- 74. Quantitative comparison of DNA ... | Nature Biotechnology methylation [https://www.nature.com/articles/nbt.3605?error=cookies_not_supported&code=68aeed35-68f6-4882-b8d0-eaa1314b37cc]
- 75. Epigenome-wide profiling of trisomy 18 specific DNA methylation signatures in first-trimester chorionic villi | Clinical Epigenetics | Full Text [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-025-02018-4]
- 76. SERS-based detection of DNA for methylation ... cancer diagnosis [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0325539]
- 77. Predicting locus-specific DNA methylation levels in cancer and paracancer tissues - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11158003/]
- 78. Identifying DNA methylation sites affecting drug response using electronic health record-derived GWAS summary statistics - PubMed [https://pubmed.ncbi.nlm.nih.gov/39670389/]
- 79. Network-based multi-omics integrative analysis methods in drug discovery: a systematic review | BioData Mining | Full Text [https://biodatamining.biomedcentral.com/articles/10.1186/s13040-025-00442-z]
- 80. For Decision Projects Matrix Research [https://www.meegle.com/en_us/topics/decision-matrix/decision-matrix-for-research-projects]
- 81. - Decision - Wikipedia matrix method [https://en.wikipedia.org/wiki/Decision-matrix_method]
- 82. The Comparison of Methods Experiment - Westgard QC [https://westgard.com/lessons/basic-method-validation/lesson23.html]