CpG Island Methylation: The Molecular Gatekeeper of Gene Silencing in Development and Disease
This comprehensive review explores the critical role of CpG island (CGI) methylation in the epigenetic regulation of gene expression.
CpG Island Methylation: The Molecular Gatekeeper of Gene Silencing in Development and Disease
Abstract
This comprehensive review explores the critical role of CpG island (CGI) methylation in the epigenetic regulation of gene expression. Targeting researchers, scientists, and drug development professionals, the article systematically covers the foundational biology of CGI promoter hypermethylation as a primary mechanism of long-term gene silencing. It details cutting-edge methodologies for detection and analysis, addresses common challenges and optimization strategies in experimental workflows, and validates findings through comparative studies of healthy and diseased states. By synthesizing current research, this article provides a holistic framework for understanding CGI methylation's implications in cancer, aging, and neurodevelopmental disorders, and highlights its potential as a target for novel therapeutics.
Decoding the Epigenetic Switch: Core Principles of CpG Island Methylation and Transcriptional Silencing
This whitepaper serves as a technical guide to defining and locating CpG islands (CGIs), a fundamental genomic element. This discussion is framed within the critical thesis that aberrant CGI methylation is a primary epigenetic mechanism driving gene silencing in diseases such as cancer, neurodegeneration, and developmental disorders. Understanding the precise definition and genomic distribution of CGIs is the essential first step for researchers investigating their methylation status and functional consequences in gene regulation and drug targeting.
Core Definition and Genomic Location
CpG islands are genomic regions with a high frequency of cytosine-guanine dinucleotides (the "CpG" site, where 'p' denotes the phosphodiester bond). Their defining characteristics are based on quantitative thresholds relative to the overall genomic background, which is depleted in CpGs due to the mutagenic potential of methylated cytosine.
Quantitative Criteria for Defining CpG Islands:
| Parameter | Classic Definition (Gardiner-Garden & Frommer, 1987) | Common Updated Criteria | Genomic Background |
|---|---|---|---|
| Length | ≥ 200 base pairs (bp) | Often ≥ 500 bp | N/A |
| GC Content | > 50% | > 55% | ~40% (mammals) |
| Observed/Expected CpG Ratio | > 0.6 | > 0.65 | ~0.2 |
CGIs are not randomly distributed. Their primary genomic locations are crucial to their regulatory function:
- Promoter-Associated CGIs: Approximately 60-70% of human gene promoters contain a CGI. These are predominantly associated with ubiquitously expressed "housekeeping" genes and many tissue-specific regulators.
- Intragenic CGIs: Located within gene bodies, often linked to alternative promoter usage or regulatory functions.
- Intergenic CGIs: Found in gene-desert regions, some may act as enhancers or insulators.
Experimental Protocols for Identification and Analysis
In SilicoIdentification and Bioinformatics Pipeline
This protocol outlines the computational identification of CGIs from sequenced genomes.
- Sequence Acquisition: Download the genomic sequence of interest (e.g., from UCSC Genome Browser, ENSEMBL) in FASTA format.
- Window Scanning: Slide a 500 bp window across the genome with a 1 bp step.
- Parameter Calculation: For each window, calculate:
- GC Content: (Number of G's + C's) / Total window length.
- Observed/Expected CpG Ratio: (Number of CpG dinucleotides) / (Number of C's * Number of G's / Window length).
- Threshold Application: Flag windows meeting criteria (e.g., length ≥500bp, GC >55%, Obs/Exp >0.65).
- Merge Adjacent Islands: Merge flagged windows separated by less than 100 bp.
- Annotation: Overlap CGI coordinates with gene annotation files (GTF/GFF) to classify location (promoter, intragenic, intergenic).
Wet-Lab Validation: Methylation-Sensitive Restriction Enzyme (MSRE) PCR
This protocol validates the methylation status of a specific predicted CGI.
- Genomic DNA Isolation: Extract high-molecular-weight DNA from target tissue/cells.
- DNA Digestion: Split DNA into three aliquots:
- Test Digest: Incubate with a methylation-sensitive enzyme (e.g., HpaII, recognition CCGG, cuts only if internal C is unmethylated).
- Control Digest: Incubate with its methylation-insensitive isoschizomer (e.g., MspI, cuts CCGG regardless of methylation).
- Undigested Control: No enzyme.
- PCR Amplification: Design primers flanking the CGI's HpaII/MspI sites. Perform PCR on all three DNA samples.
- Gel Electrophoresis: Analyze PCR products. Absence of product in HpaII digest (but presence in MspI digest) indicates methylation at those sites within the CGI.
High-Resolution Mapping: Bisulfite Sequencing
The gold standard for determining the methylation status of every cytosine within a CGI.
- Bisulfite Conversion: Treat genomic DNA with sodium bisulfite, which converts unmethylated cytosines to uracil (read as thymine in PCR), while methylated cytosines remain unchanged.
- PCR Amplification: Design primers specific for the bisulfite-converted strand of the target CGI.
- Cloning & Sequencing: Clone PCR products into a vector, sequence multiple clones to assess heterogeneity.
- Data Analysis: Align sequences to the unconverted reference. Calculate percentage methylation at each CpG site by comparing C (methylated) to T (unmethylated) calls.
Visualizing CGI Analysis Workflows
Title: In Silico CpG Island Identification Pipeline
Title: Bisulfite Sequencing Pathway for CGI Methylation
The Scientist's Toolkit: Key Research Reagent Solutions
| Research Reagent / Material | Function & Application |
|---|---|
| Sodium Bisulfite (e.g., EZ DNA Methylation Kit) | Chemical agent for deaminating unmethylated cytosine to uracil, enabling differentiation of methylation states. Essential for bisulfite sequencing. |
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, AatII) | Endonucleases that cleave DNA only at unmethylated recognition sites. Used for rapid, low-resolution validation of CGI methylation status. |
| Methylation-Insensitive Isoschizomers (e.g., MspI, AciI) | Control enzymes that cut the same recognition sequence regardless of methylation status. Paired with MSREs for validation experiments. |
| Anti-5-Methylcytosine Antibody | Antibody used for enrichment-based techniques like MeDIP (Methylated DNA Immunoprecipitation) to pull down methylated genomic fragments, including methylated CGIs. |
| PCR Primers for Bisulfite-Converted DNA | Specifically designed primers that account for C-to-T conversion to amplify target CGIs after bisulfite treatment. Critical for targeted methylation analysis. |
| Next-Generation Sequencing Kits (e.g., for WGBS) | Library preparation kits optimized for bisulfite-converted DNA, enabling genome-wide methylation profiling at single-nucleotide resolution. |
| DNA Methyltransferase Inhibitors (e.g., 5-Azacytidine, Decitabine) | Nucleoside analogs that incorporate into DNA and inhibit DNMTs, leading to global DNA hypomethylation. Used to test the functional consequence of CGI methylation on gene silencing. |
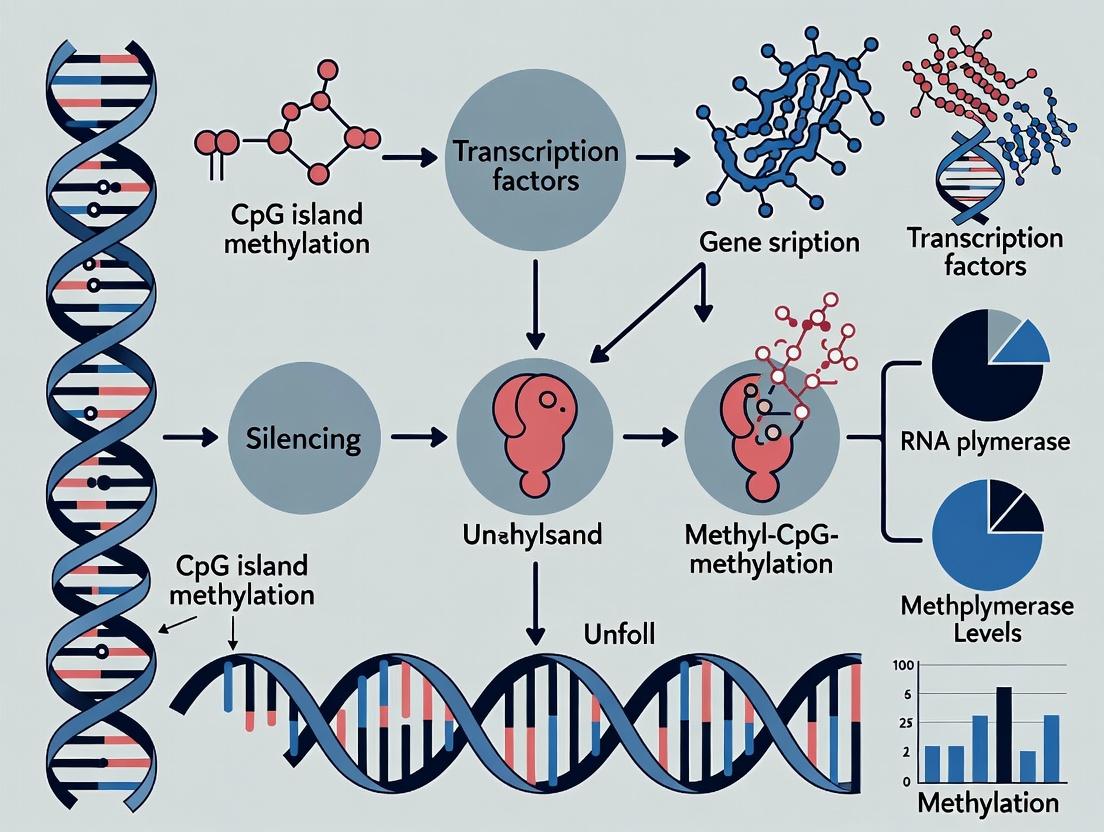
Within the broader thesis on CpG island (CGI) methylation and gene silencing, a fundamental, canonical rule has emerged: the promoters of active genes are associated with unmethylated CpG islands. This rule is a cornerstone of epigenetic regulation, linking the chemical state of DNA to transcriptional competence. Research over decades has established that aberrant methylation of these promoter CGIs is a primary mechanism of silencing tumor suppressor genes in cancer, making this field critical for understanding oncogenesis and developing epigenetic therapies. This whitepaper provides an in-depth technical guide to the core principles, evidence, and methodologies underpinning this canonical rule.
Core Mechanistic Principles
Promoter CGIs are regions of high GC density and high frequency of CpG dinucleotides. Their unmethylated state in active genes permits a permissive chromatin environment. Methylation of cytosines within CpGs (5-methylcytosine) initiates a cascade of events leading to stable gene repression.
Key Mechanistic Steps:
- Methyl-Binding Domain (MBD) Protein Recruitment: Proteins like MeCP2, MBD1, MBD2, and MBD4 bind specifically to methylated CpGs.
- Chromatin Remodeling Complex Recruitment: MBD proteins recruit histone deacetylase (HDAC) complexes and histone methyltransferases (e.g., SUV39H1).
- Histone Modification: Deacetylation of histones and methylation of histone H3 at lysine 9 (H3K9me) create a compact, transcriptionally repressive heterochromatin state.
- Polycomb Group Protein Recruitment: In some contexts, DNA methylation can facilitate the recruitment of Polycomb Repressive Complex 2 (PRC2), which catalyzes H3K27me3.
- Physical Blockade: Methylation can directly inhibit the binding of specific transcription factors (e.g., SP1, CTCF) whose recognition sequences contain CpGs.
Diagram: Methylation-Mediated Gene Silencing Pathway
Title: DNA Methylation Triggers a Repressive Chromatin Cascade
The inverse correlation between promoter CGI methylation and gene expression is supported by extensive genomic studies. The following table summarizes key quantitative findings from recent high-throughput analyses.
Table 1: Correlation Between Promoter CGI Methylation Status and Gene Expression
| Gene Category | Promoter CGI Methylation Level (%) | Median Gene Expression Level (FPKM/TPM) | Assay Used | Study (Year) |
|---|---|---|---|---|
| Highly Active Genes | 0-10 | > 50 | WGBS, RNA-seq | Schübeler et al. (2019) |
| Low/Moderate Activity | 10-30 | 5-50 | WGBS, RNA-seq | Roadmap Epigenomics (2015) |
| Silenced Genes | 70-100 | < 1 | WGBS, RNA-seq | Lister et al. (2013) |
| Tissue-Specific Genes* | >90 (inactive tissue) <10 (active tissue) | Tissue-specific pattern | RRBS, RNA-seq | Ziller et al. (2021) |
| Cancer-Suppressor Genes in Tumors | 50-100 | < 5 | Methylation arrays, qPCR | TCGA Pan-Cancer Atlas (2018) |
Example: *PAX6 promoter is unmethylated in eye tissue but hypermethylated in lymphocytes.
Table 2: Impact of Experimental CGI Demethylation on Gene Reactivation
| Treatment | Target | Result on Methylation (% reduction) | Result on Expression (fold increase) | Model System |
|---|---|---|---|---|
| 5-aza-2'-deoxycytidine (DNMTi) | Global/CGI | 20-60% global | 2-100x (locus-specific) | Cancer cell lines |
| CRISPR-dCas9-TET1 CD | Specific CGI | 40-80% at target | 5-50x | Engineered HEK293T |
| sgRNA-guided dCas9-DNMT3A | Specific CGI | 40-70% at target | 0.1-0.5x (silencing) | Engineered HEK293T |
Key Experimental Protocols
Bisulfite Sequencing for Methylation Analysis
Principle: Sodium bisulfite converts unmethylated cytosine to uracil (read as thymine after PCR), while 5-methylcytosine remains unchanged. Detailed Protocol:
- DNA Denaturation: Treat 500 ng - 1 µg of genomic DNA with NaOH (final 0.2-0.3 M) at 37°C for 15 min.
- Bisulfite Conversion: Add sodium metabisulfite (pH 5.0) and hydroquinone to the denatured DNA. Incubate in a thermal cycler: 95°C for 5 min, then 50-60°C for 4-16 hours (dark).
- Clean-up: Use commercial bisulfite cleanup kits (e.g., Zymo Research EZ DNA Methylation Kit) to desalt and remove bisulfite. Elute in 10-20 µL.
- Desulfonation: Treat with NaOH (final 0.3 M) at room temperature for 15 min, neutralize, and precipitate.
- PCR Amplification: Design primers specific for bisulfite-converted DNA (ignoring CpG sites). Use high-fidelity, bisulfite-tolerant polymerases (e.g., Taq Gold).
- Analysis: Clone PCR products and sequence 10-20 clones, or sequence directly via next-generation sequencing (Whole-Genome Bisulfite Sequencing - WGBS, or Reduced Representation Bisulfite Sequencing - RRBS). Calculate methylation percentage per CpG as (C reads / (C+T reads)).
Chromatin Immunoprecipitation (ChIP) for Correlative Histone Marks
Principle: Crosslink and shear chromatin, immunoprecipitate with antibodies against specific histone modifications, then quantify associated DNA. Detailed Protocol for H3K4me3 (Active Mark) and H3K9me3 (Repressive Mark):
- Cross-linking: Treat ~10^7 cells with 1% formaldehyde for 10 min at room temperature. Quench with glycine.
- Cell Lysis & Sonication: Lyse cells and isolate nuclei. Sonicate chromatin to shear DNA to 200-500 bp fragments. Confirm fragment size by agarose gel.
- Immunoprecipitation: Pre-clear lysate with protein A/G beads. Incubate overnight at 4°C with 2-5 µg of validated antibody (anti-H3K4me3 or anti-H3K9me3). Use normal rabbit IgG as negative control.
- Bead Capture & Washes: Capture antibody complexes with protein A/G beads. Wash sequentially with low-salt, high-salt, LiCl, and TE buffers.
- Elution & Reverse Cross-linking: Elute complexes in 1% SDS, 0.1M NaHCO3. Add NaCl to 200 mM and incubate at 65°C overnight to reverse crosslinks.
- DNA Purification: Treat with RNase A and Proteinase K. Purify DNA using phenol-chloroform extraction or spin columns.
- Analysis: Quantify precipitated DNA by qPCR with primers for the promoter CGI of interest and a control region. Express as % input or fold enrichment over IgG control.
Diagram: Integrated Workflow for Analyzing the Canonical Rule
Title: Multi-Omics Workflow to Correlate CGI Methylation and Activity
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for CGI Methylation Research
| Item Name | Supplier Examples | Function & Key Application |
|---|---|---|
| EZ DNA Methylation Kit | Zymo Research | Gold-standard for complete bisulfite conversion and clean-up of genomic DNA. |
| MethylMiner Methylated DNA Enrichment Kit | Thermo Fisher | Uses MBD-protein to enrich for methylated DNA fragments for sequencing or qPCR. |
| Magna ChIP Kit | MilliporeSigma | Complete optimized kit for Chromatin Immunoprecipitation, includes beads and buffers. |
| anti-5-Methylcytosine Antibody | Diagenode, Abcam | For immunodetection of DNA methylation (MeDIP, dot blot, immunofluorescence). |
| anti-H3K4me3 Antibody | Cell Signaling, Active Motif | Validated ChIP-grade antibody to mark active, unmethylated promoters. |
| anti-H3K9me3 Antibody | Cell Signaling, Abcam | Validated ChIP-grade antibody to mark heterochromatin linked to methylated DNA. |
| DNMT Inhibitor (5-azacytidine/Decitabine) | Selleckchem, Sigma | Small molecule inhibitors of DNA methyltransferases to induce CGI demethylation. |
| TET Enzyme (oxidation assay) | Active Motif | Recombinant enzymes to study active demethylation pathways in vitro. |
| CRISPR-dCas9-TET1/DNMT3A Systems | Addgene (Plasmids) | For targeted, locus-specific editing of methylation states without cutting DNA. |
| Infinium MethylationEPIC BeadChip | Illumina | Array-based platform for profiling methylation at >850,000 CpG sites genome-wide. |
Within the broader thesis of CpG island (CGI) methylation and gene silencing research, the phenomenon of aberrant CGI hypermethylation represents a critical paradox. Canonically, CpG islands in gene promoters are protected from methylation, ensuring active gene expression. "Breaking this rule" is a hallmark of cancer and other diseases, leading to the silencing of tumor suppressor genes and genomic instability. This whitepaper provides an in-depth technical analysis of the current understanding of the mechanisms and triggers that lead to this pathogenic state, intended for researchers and drug development professionals.
Core Mechanisms of Aberrant CGI Hypermethylation
The initiation and maintenance of aberrant hypermethylation are governed by interconnected mechanisms disrupting the normal epigenetic landscape.
Dysregulation of the DNA Methylation Machinery
Aberrant activity of DNA methyltransferases (DNMTs) is a primary driver. While DNMT1 is crucial for maintenance methylation, DNMT3A and DNMT3B perform de novo methylation. In cancer, overexpression of these enzymes, particularly DNMT3B, is frequently observed. Recent studies highlight the role of somatic mutations in DNMT3A (e.g., R882H) that alter enzyme activity and specificity, potentially contributing to aberrant methylation patterns in hematological malignancies.
Histone Modification Crosstalk
A well-established mechanism involves the polycomb repressive complex 2 (PRC2). H3K27me3, deposited by PRC2, can recruit DNMTs, creating a bridge from facultative heterochromatin to a more stable, DNA methylation-based silencing state. This "histone code-guided DNA methylation" is a key pathway for initiating hypermethylation at specific loci.
Transcription Factor-Mediated Protection Loss
In normal cells, transcription factors (TFs) like SP1 and MYC bind to unmethylated CGI promoters, blocking DNMT access. Their loss of binding due to mutation, decreased expression, or competitive displacement allows the de novo methylation machinery to target the now-vulnerable CGI.
Disruption of Protective Demethylation Pathways
Active demethylation, mediated by TET enzymes oxidizing 5mC to 5hmC, 5fC, and 5caC, protects CGIs. Mutations in TET2, IDH1/2 (which produce the oncometabolite 2-HG inhibiting TETs), or depletion of ascorbate (a TET cofactor) disrupt this protective cycle, leading to methylation buildup.
Diagram 1: Logical flow from trigger to disease.
Key Triggers and Initiating Events
Genetic Alterations
Mutations in genes encoding epigenetic regulators (DNMT3A, TET2, IDH1/2) are direct triggers. Furthermore, chromosomal translocations can bring CGI promoters into proximity with repressive genomic compartments or methylated regions.
Aging
Aging is the most potent physiological trigger, associated with a progressive increase in CGI methylation at specific loci, a process accelerated and dysregulated in cancer.
Environmental and Lifestyle Exposures
- Chronic Inflammation: Inflammatory cytokines (e.g., IL-6) can upregulate DNMT expression and activity.
- Tobacco Smoke: Contains pro-methylation agents like reactive aldehydes.
- Dietary Factors: Folate deficiency can alter the SAM/SAH methylation balance.
Viral Integration
Viruses like HPV and HBV can induce localized hypermethylation of integrated host gene promoters as part of their oncogenic strategy.
Table 1: Prevalence of Epigenetic Alterations in Selected Cancers
| Cancer Type | Gene Frequently Hypermethylated | Approximate Frequency | Associated Trigger/Mutation |
|---|---|---|---|
| Colorectal Cancer | MLH1 (Mismatch Repair) | 10-15% | Sporadic MSI; linked to aging & inflammation |
| Glioblastoma | MGMT (DNA Repair) | ~40% | Response predictor to temozolomide |
| Acute Myeloid Leukemia | Multiple CGIs (CIMP phenotype) | 15-20% | High correlation with IDH1/2 or TET2 mutations |
| Breast Cancer | BRCA1 (DNA Repair) | 10-15% in sporadic cases | Associated with loss of transcription factor binding |
Table 2: Key Enzymatic Activities in CGI Methylation Regulation
| Enzyme/Complex | Primary Function | Effect on CGI Methylation | Common Aberration in Disease |
|---|---|---|---|
| DNMT3A/3B | De novo DNA methylation | Increase | Overexpression, Gain-of-function mutations |
| TET2 | 5mC Oxidation (initiates demethylation) | Decrease | Loss-of-function mutations, Inhibition by 2-HG |
| PRC2 (EZH2) | Deposits H3K27me3 | Facilitates Increase | Overexpression, Recruits DNMTs |
| DNMT1 | Maintenance DNA methylation | Sustains Increase | Overexpression, Altered targeting |
Experimental Protocols for Key Investigations
Protocol: Genome-wide Profiling of 5mC/5hmC in CGI Regions
Objective: To simultaneously map 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) at base resolution in CGIs. Method: Oxidative Bisulfite Sequencing (oxBS-Seq) combined with CGI capture.
- DNA Extraction & Quality Control: Isolate high-molecular-weight DNA. Verify integrity via agarose gel electrophoresis.
- Oxidative Treatment (oxBS): Split DNA into two aliquots.
- oxBS-treated: Incubate with KRuO4 to oxidize 5hmC to 5fC.
- BS-only control: Treated with standard bisulfite.
- Bisulfite Conversion: Use the EZ DNA Methylation-Lightning Kit (Zymo Research) on both aliquots. 5mC and 5hmC resist conversion in the BS-only sample; only 5mC resists in the oxBS sample.
- CGI Enrichment: Use the SeqCap Epi CpGiant Enrichment Kit (Roche) following bisulfite-converted DNA library preparation.
- High-Throughput Sequencing & Analysis: Sequence on an Illumina platform. Align reads to a bisulfite-converted reference genome. 5hmC level = (BS methylation % - oxBS methylation %).
Protocol: Assessing the Functional Consequence of CGI Methylation
Objective: To determine if CGI hypermethylation directly causes gene silencing. Method: In vitro Methylation and Reporter Assay.
- Reporter Construct Cloning: Clone the candidate CGI/promoter sequence upstream of a luciferase reporter gene (e.g., pGL4-basic vector).
- In vitro Methylation: Treat purified plasmid DNA with excess M.SssI (CpG Methyltransferase, NEB) in the presence of S-adenosylmethionine (SAM) to achieve complete in vitro CpG methylation. Verify methylation by HpaII/MspI restriction digest.
- Cell Transfection: Co-transfect methylated and unmethylated reporter plasmids, along with a Renilla control plasmid for normalization, into relevant cell lines (e.g., HEK293 or a matched cancer cell line).
- Dual-Luciferase Assay: After 48h, lyse cells and measure Firefly and Renilla luciferase activity using the Dual-Luciferase Reporter Assay System (Promega). Calculate the normalized ratio (Firefly/Renilla). Silencing is indicated by a significant reduction in luminescence from the methylated vs. unmethylated plasmid.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Aberrant CGI Hypermethylation Research
| Item (Example Vendor) | Function in Research | Application Notes |
|---|---|---|
| M.SssI CpG Methyltransferase (NEB) | Catalyzes in vitro methylation of all CpG sites. | Used for functional validation in reporter assays or creating fully methylated DNA controls. |
| 5-Aza-2'-deoxycytidine (Decitabine) (Sigma-Aldrich) | DNMT inhibitor; incorporates into DNA, trapping DNMTs and promoting their degradation. | Positive control for demethylation experiments; used clinically in MDS/AML. |
| Recombinant Human TET2 Catalytic Domain (Active Motif) | Enzyme for in vitro 5mC oxidation assays. | Useful for studying demethylation kinetics or generating 5hmC/5fC/5caC standards. |
| EZH2 Inhibitor (GSK126, Cayman Chemical) | Selective small-molecule inhibitor of PRC2's H3K27 methyltransferase activity. | Probes the role of H3K27me3 in facilitating subsequent DNA methylation. |
| Anti-5hmC Antibody (Clone RM236, RevMAb) | Highly specific monoclonal antibody for immuno-detection of 5-hydroxymethylcytosine. | Used in dot-blot, immunofluorescence, or hMeDIP-seq to assess active demethylation states. |
| EZ DNA Methylation-Lightning Kit (Zymo Research) | Rapid bisulfite conversion of DNA for downstream methylation analysis. | Industry standard for preparing samples for pyrosequencing, MSP, or bisulfite sequencing. |
| CpG Methylase M.CviPI (NEB) | Methylates GpC sites (not CpG). | Critical control enzyme for "Methylase Accessibility Assay" to study chromatin structure independent of CpG methylation. |
Signaling Pathways in Inflammation-Induced Hypermethylation
Diagram 2: Inflammation pathway driving CGI hypermethylation.
Within the broader context of CpG island methylation and gene silencing research, this whitepaper details the mechanistic link between DNA methylation, methyl-CpG-binding domain (MBD) proteins, and histone deacetylase (HDAC) complexes. This epigenetic silencing cascade is fundamental to gene regulation, development, and disease, making it a critical target for therapeutic intervention.
Gene silencing initiated by CpG island methylation is not mediated by methylated DNA alone. The repressive signal is translated into a transcriptionally inactive chromatin state through a two-step recruitment process: 1) the binding of MBD proteins to symmetrically methylated CpG dinucleotides, and 2) the subsequent recruitment of HDAC-containing co-repressor complexes that remodel chromatin.
Core Molecular Players
The Methyl-CpG-Binding Domain (MBD) Family
MBD proteins act as interpreters of the DNA methylation mark. The canonical family includes MeCP2, MBD1, MBD2, MBD3, and MBD4. They share a conserved MBD that selectively recognizes 5-methylcytosine.
Histone Deacetylase (HDAC) Complexes
HDACs (primarily Class I HDACs 1, 2, and 3 within this context) remove acetyl groups from histone lysine tails, leading to chromatin compaction and transcriptional repression. They are typically part of large multi-protein complexes like Sin3, NuRD, and CoREST.
Mechanistic Recruitment Pathways
The recruitment process follows a defined molecular logic, as illustrated below.
Diagram Title: DNA Methylation to Chromatin Silencing Pathway
Quantitative Analysis of Key Interactions
The affinity and functional outcomes of MBD-HDAC recruitment vary across family members.
Table 1: Characteristics of Major MBD Proteins in HDAC Recruitment
| MBD Protein | Primary HDAC Complex Recruited | Binding Affinity for Methylated DNA (Kd approx.) | Key Functional Domains |
|---|---|---|---|
| MeCP2 | Sin3A | 1-10 nM | MBD, TRD (Transcriptional Repression Domain) |
| MBD2 | NuRD (via MBD3) | 5-20 nM | MBD, GR (Glycine-Arginine rich) |
| MBD1 | Sin3A, CAF-1 | 10-50 nM | MBD, CXXC3, TRD |
| MBD3 | NuRD (Integral Component) | Does not bind methylated DNA | MBD (non-binding) |
Table 2: HDAC Complexes in Methylation-Dependent Silencing
| HDAC Complex | Core HDACs | Associated MBD Proteins | Key Additional Components |
|---|---|---|---|
| Sin3 | HDAC1, HDAC2 | MeCP2, MBD1 | SAP18, SAP30, RbAp46/48 |
| NuRD | HDAC1, HDAC2 | MBD2, MBD3 (integral) | MTA1/2/3, CHD3/4, RbAp46/48 |
| CoREST | HDAC1, HDAC2 | MeCP2 (context-specific) | RCOR1, LSD1, BRAF35 |
Experimental Protocols for Investigating the Machinery
Protocol: Co-Immunoprecipitation (Co-IP) for MBD-HDAC Complex Analysis
Objective: To validate physical interaction between a specific MBD protein and an HDAC complex.
Methodology:
- Cell Lysis: Harvest HeLa or HEK293T cells (or relevant cell line). Lyse in IP lysis buffer (25mM Tris-HCl pH 7.4, 150mM NaCl, 1% NP-40, 1mM EDTA, 5% glycerol) supplemented with protease and HDAC inhibitors (e.g., sodium butyrate) for 30 min on ice.
- Pre-clearing: Incubate lysate with Protein A/G beads for 1 hour at 4°C to reduce non-specific binding.
- Immunoprecipitation: Incubate pre-cleared lysate with 2-5 µg of antibody against the target MBD protein (e.g., anti-MeCP2) or control IgG overnight at 4°C with gentle rotation.
- Bead Capture: Add Protein A/G agarose beads and incubate for 2 hours.
- Washing: Pellet beads and wash 4x with lysis buffer.
- Elution: Elute bound proteins by boiling in 2X Laemmli sample buffer.
- Analysis: Resolve proteins by SDS-PAGE and perform Western blotting for the MBD protein, HDAC1/2, and complex-specific subunits (e.g., Sin3A).
Protocol: Chromatin Immunoprecipitation (ChIP) Sequential (Re-ChIP)
Objective: To demonstrate co-occupancy of an MBD protein and an HDAC on the same methylated genomic locus.
Methodology:
- First Crosslinking & ChIP: Crosslink cells with 1% formaldehyde for 10 min. Quench with glycine. Sonicate chromatin to 200-500 bp fragments. Perform standard ChIP using an anti-MBD antibody (e.g., anti-MBD2).
- Elution for Re-ChIP: Elute the immune complexes from the first ChIP beads not with SDS buffer, but with 10mM DTT for 30 min at 37°C.
- Dilution & Second ChIP: Dilute the eluate 1:50 in Re-ChIP buffer (1% Triton X-100, 2mM EDTA, 150mM NaCl, 20mM Tris-HCl pH 8.1). Perform a second ChIP using an antibody against an HDAC or complex component (e.g., anti-HDAC1).
- DNA Recovery: Reverse crosslinks, purify DNA, and analyze by qPCR targeting a known methylated/silenced gene promoter and a control active region.
Protocol: In Vitro Histone Deacetylase Activity Assay
Objective: To measure HDAC activity recruited by an MBD protein in a purified system.
Methodology:
- Reconstitute Complex: Incubate recombinant MBD protein (e.g., MeCP2) with immunopurified Sin3/HDAC complex.
- Assay Setup: Use a fluorogenic HDAC substrate (e.g., acetylated Lysine substrate). In a 96-well plate, mix substrate with the protein complex in HDAC assay buffer.
- Reaction & Developer: Incubate for 1-2 hours at 37°C. Stop the reaction and add developer to cleave the deacetylated product, releasing a fluorescent signal.
- Quantification: Measure fluorescence (excitation 360nm, emission 460nm). Compare activity with controls (no MBD protein, HDAC inhibitor control like Trichostatin A).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Studying Methylation-Dependent Silencing
| Reagent / Material | Function & Application | Example Product / Target |
|---|---|---|
| DNA Methyltransferase Inhibitor | Demethylates genome to test causality of methylation in silencing. | 5-Aza-2'-deoxycytidine (Decitabine) |
| HDAC Inhibitors | Blocks HDAC activity to test functional outcome of recruitment. | Trichostatin A (TSA), Suberoylanilide Hydroxamic Acid (SAHA/Vorinostat) |
| MBD-Specific Antibodies | For IP, ChIP, and WB to detect/probe MBD proteins. | Anti-MeCP2, Anti-MBD2, Anti-MBD1 (validated for ChIP-grade) |
| HDAC/Complex Antibodies | For detecting co-repressor complexes. | Anti-HDAC1, Anti-Sin3A, Anti-MTA2 (NuRD) |
| Bisulfite Conversion Kit | Maps DNA methylation patterns at CpG islands of silenced genes. | EZ DNA Methylation-Lightning Kit |
| Fluorogenic HDAC Assay Kit | Quantifies HDAC activity in vitro or from immunoprecipitated complexes. | HDAC-Glo I/II Assay |
| Methylated DNA Probes | For pull-down assays to study MBD protein recruitment. | Biotinylated methylated CpG oligonucleotides |
| Recombinant MBD Proteins | For in vitro binding and recruitment studies. | Recombinant human MeCP2, MBD2 protein |
Therapeutic Implications and Drug Development
Understanding this recruitment machinery provides direct targets for epigenetic therapy. DNMT inhibitors (e.g., Azacitidine) and HDAC inhibitors (e.g., Romidepsin) are approved for hematological malignancies. Current research focuses on developing protein-protein interaction inhibitors to disrupt specific MBD-co-repressor binding, aiming for greater specificity.
Concluding Remarks
The sequential recruitment of MBD proteins and HDAC complexes forms the core effector mechanism of DNA methylation-mediated gene silencing. Disrupting this interface holds significant promise for reversing pathological epigenetic states in cancer and neurological disorders.
Within the broader thesis on CpG island (CGI) methylation as a central mechanism for heritable gene silencing, this whitepaper examines its pivotal biological roles. CGI methylation is not a default state but a highly regulated process, with its establishment and maintenance being crucial for three fundamental epigenetic phenomena: X-chromosome inactivation (XCI), genomic imprinting, and cellular differentiation. Understanding the precise timing, targeting, and functional consequences in these contexts is essential for unraveling developmental biology and disease etiology.
CGI Methylation in X-Chromosome Inactivation
XCI ensures dosage compensation in female mammals by silencing one of the two X chromosomes. While the initial silencing is orchestrated by the long non-coding RNA Xist and its associated repressive complexes, CGI methylation serves as the long-term, stable lock for maintaining the inactive state (Xi) through cell divisions.
- Key Target: The promoter CGI of genes on the Xi.
- Timing: Methylation is a late event, consolidating after gene silencing has been initiated by histone modifications (e.g., H3K27me3).
Table 1: Key Quantitative Data in X-Inactivation
| Metric | Value/Observation | Technical Note |
|---|---|---|
| Percentage of genes on Xi with methylated promoter CGIs | ~85% | Remaining genes escape XCI; their CGIs stay hypomethylated. |
| Typical methylation level at silenced CGI promoters on Xi | >70% | Measured via bisulfite sequencing in clonal cell populations. |
| Timeframe for CGI methylation establishment post-Xist coating | Weeks (in vitro differentiation models) | Consolidation occurs long after transcriptional shutdown. |
Experimental Protocol: Analyzing Allele-Specific Methylation on Xi
- Method: Allele-Specific Bisulfite Sequencing (BS-seq).
- Steps:
- Cell Preparation: Use hybrid mouse cells (e.g., Mus musculus x Mus castaneus) or human cells with heterozygous SNPs.
- Nucleic Acid Extraction: Isolate genomic DNA.
- Bisulfite Conversion: Treat DNA with sodium bisulfite, converting unmethylated cytosine to uracil (reads as thymine in sequencing), while methylated cytosine remains unchanged.
- PCR Amplification: Amplify target CGI regions using primers designed for bisulfite-converted DNA.
- Sequencing & Analysis: Perform deep sequencing. Align reads to a reference genome and discriminate alleles using known SNPs. Methylation calls per CpG are derived, allowing comparison between the active (Xa) and inactive (Xi) alleles.
CGI Methylation in Genomic Imprinting
Genomic imprinting results in parent-of-origin-specific monoallelic expression. Differentially methylated regions (DMRs), often encompassing CGIs, are established in the germline and serve as the primary imprinting control marks.
- Key Target: Germline DMRs (gDMRs), which are often intergenic or intronic CGIs.
- Timing: Established during gametogenesis, maintained post-fertilization throughout somatic development.
Table 2: Key Quantitative Data in Genomic Imprinting
| Metric | Value/Observation | Technical Note |
|---|---|---|
| Number of confirmed imprinted human genes | ~150-200 | Defined by the presence of a gDMR. |
| Methylation difference at a canonical gDMR (e.g., IGF2/H19 ICR) | ~50% (Methylated Allele: 90-100%; Unmethylated Allele: 0-10%) | Idealized data; measured via pyrosequencing or BS-seq. |
| Size of a typical imprinting control region (ICR) | 1-5 kb | Often spans a CGI. |
Experimental Protocol: Identifying Novel Imprinted Loci via Methylome Analysis
- Method: Whole-Genome Bisulfite Sequencing (WGBS) of reciprocal hybrids or tissues with parthenogenetic/androgenetic origin.
- Steps:
- Sample Collection: Generate or obtain biological samples with distinct parental genomes (e.g., mouse embryos from reciprocal crosses).
- WGBS Library Prep: Fragment genomic DNA, perform bisulfite conversion, and prepare sequencing libraries with appropriate adapters.
- High-Throughput Sequencing: Sequence on platforms like Illumina NovaSeq.
- Bioinformatic Pipeline: Align reads to a bisulfite-converted reference genome. Identify genomic positions where methylation levels are consistently ~50% overall but show allele-specific patterns when parental SNPs are considered, indicating a gDMR.
CGI Methylation in Cellular Differentiation
During lineage commitment, de novo methylation of CGI-associated promoters permanently silences pluripotency and alternative lineage genes, while housekeeping and lineage-specific gene CGIs remain protected.
- Key Target: Developmental gene promoter CGIs.
- Timing: Occurs during gastrulation and subsequent tissue specification.
Table 3: Key Quantitative Data in Cellular Differentiation
| Metric | Value/Observation | Technical Note |
|---|---|---|
| Estimated CGI promoters gaining methylation during human somatic differentiation | ~20-30% | Varies significantly by tissue lineage. |
| Methylation increase at a silenced pluripotency gene promoter (e.g., OCT4/POU5F1) | From <10% to >80% | Measured during in vitro differentiation of hESCs. |
| Number of de novo methyltransferases involved | 2 (DNMT3A & DNMT3B, with cofactor DNMT3L) | DNMT1 maintains the pattern. |
Experimental Protocol: Tracking Methylation Dynamics During Differentiation
- Method: Time-course Methylated DNA Immunoprecipitation Sequencing (MeDIP-seq) or Reduced Representation Bisulfite Sequencing (RRBS).
- Steps:
- Time-Course Design: Differentiate pluripotent stem cells (e.g., ESCs) into a target lineage. Harvest cells at days 0, 2, 5, 10, etc.
- DNA Extraction & Processing: Isolate genomic DNA. For RRBS, digest with MspI (cuts CCGG) to enrich for CpG-rich regions.
- Immunoprecipitation (MeDIP): Fragment DNA, denature, and incubate with an antibody specific to 5-methylcytosine. Pull down methylated fragments.
- Library Prep & Sequencing: Prepare sequencing libraries from immunoprecipitated DNA (MeDIP) or bisulfite-converted RRBS fragments.
- Analysis: Map reads to the genome. For MeDIP, analyze enrichment peaks over promoter CGIs. For RRBS, calculate methylation percentages. Identify loci where methylation increases progressively over time.
Visualizations
Title: Sequential Steps in X-Chromosome Inactivation
Title: Lifecycle of a Genomic Imprint
Title: CGI Methylation Locks in Cell Fate
The Scientist's Toolkit: Key Reagents & Materials
| Item | Function in CGI Methylation Research |
|---|---|
| Sodium Bisulfite | Chemical reagent that converts unmethylated cytosine to uracil for sequencing-based methylation analysis. |
| Anti-5-Methylcytosine Antibody | For immunoprecipitation-based methods (MeDIP, mDIP) to enrich methylated DNA fragments. |
| DNMT Inhibitors (e.g., 5-Azacytidine, Decitabine) | Nucleoside analogs that incorporate into DNA and trap DNA methyltransferases, leading to global demethylation; used for functional studies. |
| M.SssI Methyltransferase | Bacterial enzyme that methylates all CpG sites in vitro; used as a positive control or for spiking experiments. |
| PCR Primers for Bisulfite-Converted DNA | Specifically designed to amplify sequences post-bisulfite treatment, ignoring methylation status. |
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII) | Enzymes that cut only unmethylated CCGG sites; used in assays like RLGS or MS-RE-PCR. |
| Targeted Bisulfite Sequencing Kits (e.g., PyroMark, EpiTYPER) | Commercial systems for quantitative, high-throughput methylation analysis of specific loci. |
| Stable Isotope-Labeled Methionine (e.g., 13C-Met) | Allows tracking of methyl group incorporation into DNA via mass spectrometry (stable isotope tracing). |
| dCas9-DNMT3A/3L Fusion Constructs | For targeted methylation of specific CGI sequences in epigenetic editing experiments. |
| TET Enzyme Catalytic Domain Constructs | For targeted demethylation of methylated CGIs to assess functional consequences. |
The epigenetic silencing of tumor suppressor genes (TSGs) via the hypermethylation of CpG islands (CGIs) in their promoter regions is a well-established hallmark of cancer. This whitepaper situates this mechanism within the broader thesis of CpG island methylation research, which seeks to understand the precise triggers, patterns, and consequences of this aberrant epigenetic mark. For drug development professionals, this represents a prime target for epigenetic therapies aimed at reversing silencing and restoring TSG function.
Core Mechanism and Quantitative Data
Hypermethylation at CGI promoters leads to a repressive chromatin state. Methyl-CpG-binding domain (MBD) proteins recruit histone deacetylases (HDACs) and histone methyltransferases (HMTs), leading to histone H3 deacetylation and increased H3K9me3/H3K27me3 marks. This closed chromatin structure blocks transcription factor binding and RNA polymerase II recruitment, permanently silencing TSGs critical for cell cycle control, DNA repair, and apoptosis.
Table 1: Frequently Inactivated TSGs via CGI Hypermethylation in Human Cancers
| Tumor Suppressor Gene | Primary Function | Key Cancer Types with Frequent Promoter Hypermethylation | Approximate Frequency Range |
|---|---|---|---|
| CDKN2A (p16INK4a) | Cell cycle inhibitor (G1/S checkpoint) | Colorectal, Lung, Pancreatic, Glioblastoma | 20-80% depending on type |
| BRCA1 | DNA damage repair (Homologous recombination) | Breast, Ovarian (sporadic), Triple-negative Breast Cancer | 10-30% |
| MLH1 | DNA mismatch repair | Colorectal (sporadic MSI-H), Endometrial | 10-20% in sporadic MSI-H CRC |
| RASSF1A | Microtubule stability, apoptosis | Lung, Breast, Renal, Neuroblastoma | 40-90% |
| MGMT | DNA repair (alkylation damage) | Glioblastoma, Colorectal | 20-50% in GBM |
| VHL | Hypoxia response regulation | Renal Cell Carcinoma (sporadic) | 5-20% |
| APC | Wnt signaling regulator | Colorectal, Gastric | ~5% (often mutated, but methylated in some subsets) |
Table 2: Technologies for Quantifying CGI Methylation
| Technology | Principle | Application | Sensitivity | Throughput |
|---|---|---|---|---|
| Bisulfite Sequencing (WGBS, RRBS) | Converts unmethylated C to U; methylated C remains. Sequencing reveals methylated positions. | Genome-wide or reduced representation methylation profiling. | High (single-base, can detect low allele frequency) | Low to Medium |
| Methylation-Specific PCR (MSP) | Bisulfite-treated DNA amplified with primers specific for methylated or unmethylated sequences. | Targeted, clinical screening of known CGI regions. | High (can detect <0.1% methylated alleles) | High |
| Pyrosequencing | Quantitative sequencing-by-synthesis of bisulfite-converted DNA. | Targeted, absolute quantification of methylation percentage at specific CpGs. | High (quantitative, ~5% sensitivity) | Medium |
| Methylation BeadChip (e.g., EPIC) | Bead-based array hybridizing bisulfite-converted DNA to probe sets. | Genome-wide profiling of predefined CpG sites (850,000+ sites). | Medium | Very High |
| MeDIP-seq / MBD-seq | Immunoprecipitation of methylated DNA with anti-5mC antibody or MBD proteins, followed by sequencing. | Genome-wide enrichment-based methylation analysis. | Lower resolution (~100bp regions) | Medium |
Experimental Protocols
Protocol: Combined Bisulfite Restriction Analysis (COBRA) for Targeted CGI Methylation Validation
Objective: To quantitatively assess the methylation status of a specific CpG island following a genome-wide screen.
Materials:
- Sodium bisulfite conversion kit (e.g., EZ DNA Methylation-Lightning Kit)
- PCR reagents and primers designed for bisulfite-converted DNA (flanking a restriction site containing CpG)
- Restriction enzyme (BstUI (CG^CG) or TaqI (T^CGA) are common for bisulfite-converted sequences)
- Agarose gel electrophoresis system
- Densitometry software
Procedure:
- Bisulfite Conversion: Treat 500 ng of genomic DNA from tumor and matched normal tissue using the kit. This converts unmethylated cytosines to uracil, leaving 5-methylcytosines unchanged.
- PCR Amplification: Design primers that amplify a 200-300bp region of the bisulfite-converted CGI of interest. The amplicon must contain at least one restriction site that is retained only if the CpG within the site was originally methylated (e.g., BstUI site "CGCG" becomes "CGCG" if methylated, or "UGCG"/"TGCG" if unmethylated and thus unconvertible/unrecognizable).
- Restriction Digestion: Purify the PCR product. Split into two aliquots: one digested with the methylation-sensitive enzyme (e.g., BstUI) and one undigested control. Incubate at enzyme-optimal temperature for 4 hours.
- Electrophoresis & Analysis: Run digested and undigested products on a 2-3% agarose gel. The undigested product shows the full-length amplicon. The digested product shows:
- Unmethylated DNA: No cutting, single band.
- Fully Methylated DNA: Complete cutting, two smaller bands.
- Heterogeneously Methylated DNA: A mixture of all three bands.
- Quantification: Use gel densitometry to calculate the percentage of methylated alleles: (Intensity of cut bands) / (Total intensity of all bands) x 100.
Protocol: Chromatin Immunoprecipitation (ChIP) for Assessing Repressive Histone Marks
Objective: To confirm the establishment of a repressive chromatin state following CGI hypermethylation at a specific TSG promoter.
Materials:
- Crosslinking reagent (1% formaldehyde)
- Cell lysis buffers (SDS Lysis Buffer, IP Buffer)
- Sonicator for chromatin shearing
- Antibodies: anti-H3K9me3, anti-H3K27me3, anti-H3 (control), normal rabbit IgG (negative control)
- Protein A/G magnetic beads
- PCR or qPCR reagents for target promoter region and a control non-methylated region.
Procedure:
- Crosslinking & Harvesting: Fix cells in culture with 1% formaldehyde for 10 min at room temperature. Quench with glycine. Harvest cells and wash with cold PBS.
- Cell Lysis & Sonication: Lyse cells in SDS Lysis Buffer. Sonicate chromatin to shear DNA to fragments of 200-1000 bp. Centrifuge to clear debris.
- Immunoprecipitation: Dilute chromatin supernatant in IP Buffer. Aliquot for input control and IP samples. Add appropriate antibody to each IP sample (e.g., anti-H3K9me3). Incubate overnight at 4°C with rotation.
- Bead Capture & Washes: Add Protein A/G beads, incubate, and wash sequentially with low salt, high salt, LiCl, and TE buffers.
- Elution & Reverse Crosslinking: Elute chromatin from beads. Reverse crosslinks for both IP and input samples at 65°C overnight.
- DNA Purification & Analysis: Purify DNA. Analyze by qPCR using primers specific for the TSG promoter CGI and a control gene region (e.g., GAPDH promoter). Enrichment is calculated as % Input or fold-change over IgG control.
Visualizations
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for CGI Hypermethylation Research
| Reagent / Kit | Primary Function | Key Consideration for Selection |
|---|---|---|
| Sodium Bisulfite Conversion Kits (e.g., EZ DNA Methylation, InnuConvert) | Converts unmethylated cytosine to uracil for downstream methylation analysis. | Conversion efficiency, DNA input requirements, speed, and compatibility with degraded DNA (FFPE). |
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, BstUI, Acil) | Cut only at unmethylated recognition sites. Used in COBRA, HELP-seq, etc. | Specificity, star activity, buffer compatibility with PCR products. |
| Anti-5-Methylcytosine (5mC) Antibodies | For immunoprecipitation-based methods (MeDIP) or immunofluorescence. | Clonality, specificity (no cross-reactivity to 5hmC), ChIP-grade validation. |
| MBD-Based Capture Kits (e.g., MethylMiner, MethylCap) | Uses recombinant MBD proteins to isolate methylated DNA fragments. | Binding affinity, fragment size selection, background (non-specific binding). |
| DNMT Inhibitors (e.g., 5-Azacytidine, Decitabine) | Hypomethylating agents used in vitro to reverse CGI hypermethylation and test functional reactivation. | Cytotoxicity, concentration, duration of treatment for optimal demethylation vs. cell death. |
| Validated Primers for Bisulfite Sequencing/Pyrosequencing | Target-specific amplification of bisulfite-converted DNA. | Must be designed specifically for converted sequence (no CpGs in primer if possible), specificity, amplicon size. |
| qPCR Assays for Methylation Analysis (e.g., MethylLight, MS-HRM) | Quantitative, high-throughput detection of methylation at specific loci. | Probe specificity (methylated vs. unmethylated), sensitivity, multiplexing capability. |
| ChIP-Validated Antibodies for Repressive Marks (e.g., anti-H3K9me3, anti-H3K27me3) | To correlate DNA methylation with histone modification status via ChIP. | Specificity validated by knockout/knockdown cells, lot-to-lot consistency, high signal-to-noise in ChIP. |
From Bench to Bedside: Advanced Techniques for Profiling CGI Methylation in Research and Diagnostics
Within the central thesis of CpG island methylation and gene silencing research, understanding the precise methylation status of cytosines is paramount. Bisulfite conversion of DNA remains the foundational chemical reaction enabling this interrogation, transforming unmethylated cytosines to uracils while leaving methylated cytosines intact. This whitepaper provides an in-depth technical guide to three gold-standard, bisulfite-dependent methodologies: Whole-Genome Bisulfite Sequencing (WGBS), Reduced Representation Bisulfite Sequencing (RRBS), and Pyrosequencing.
Core Principles and Quantitative Comparison
Bisulfite conversion exploits the differential deamination rates of cytosine and 5-methylcytosine under acidic conditions. Subsequent PCR amplification converts uracils to thymines, creating sequence polymorphisms that can be detected by sequencing or quantitative assays.
Table 1: Comparative Analysis of Key Bisulfite-Based Methods
| Feature | Whole-Genome Bisulfite Sequencing (WGBS) | Reduced Representation Bisulfite Sequencing (RRBS) | Pyrosequencing |
|---|---|---|---|
| Genome Coverage | >90% of CpGs (theoretical) | ~3-5 million CpGs, enriched in CpG islands and promoters | Targeted (typically < 200bp amplicon) |
| Resolution | Single-base pair | Single-base pair | Single-base pair (averaged per CpG unit) |
| Typical Input DNA | 50-200 ng (standard), <10 ng (ultra-low input) | 10-100 ng | 10-50 ng of bisulfite-converted DNA |
| Key Advantage | Comprehensive, hypothesis-free, detects non-CpG methylation | Cost-effective, high coverage of regulatory regions, simpler data analysis | Highly quantitative, accurate, rapid, no cloning needed |
| Primary Limitation | High cost, complex bioinformatics, high DNA input for full coverage | Bias towards high-CpG-density regions, misses low-CpG regions | Low multiplexing, short read length, requires prior target selection |
| Best For | Discovery-based studies, imprinted genes, non-CpG methylation, novel biomarker identification | Large-scale epigenotyping, cancer studies focusing on promoter hypermethylation | Validation of NGS data, clinical biomarker quantification, longitudinal studies |
Detailed Experimental Protocols
Protocol 1: Sodium Bisulfite Conversion (Common Initial Step)
- Principle: Denatured DNA is treated with sodium bisulfite, which sulfonates unmethylated cytosine. Subsequent desulfonation under alkaline conditions yields uracil. Methylated cytosine is unreactive.
- Key Steps:
- Denaturation: 1-2 µg of genomic DNA in a volume of 20 µL is denatured by adding 2.2 µL of 3M NaOH and incubating at 37°C for 15 minutes.
- Sulfonation: 208 µL of a freshly prepared 10mM hydroquinone solution and 1.2 mL of 3.6M sodium bisulfite (pH 5.0) are added. The mixture is incubated under mineral oil in the dark (16-20 hours at 55°C).
- Desalting: DNA is bound to a silica membrane column (e.g., using a commercial kit), washed, and desulfonated on-column with 0.3M NaOH for 15 minutes at room temperature.
- Neutralization & Elution: The column is neutralized with a buffer, washed, and DNA is eluted in 10-20 µL of low-EDTA TE buffer or nuclease-free water. Converted DNA is stored at -80°C.
Protocol 2: Reduced Representation Bisulfite Sequencing (RRBS)
- Restriction Digestion: 10-100 ng of genomic DNA is digested with the CpG-methylation insensitive restriction enzyme MspI (cuts CCGG), which enriches for CpG-rich fragments.
- End-Repair & A-Tailing: Digested fragments undergo end-repair and 3'-adenylation to prepare them for adapter ligation.
- Adapter Ligation: Methylated adapters (compatible with bisulfite sequencing) are ligated to the fragments.
- ͏Size Selection: Fragments in the 40-220 bp range (containing CpG islands) are selected via gel extraction or bead-based methods.
- Bisulfite Conversion: The size-selected library is subjected to sodium bisulfite conversion as described in Protocol 1.
- PCR Amplification: The converted library is amplified with a low number of PCR cycles using primers complementary to the adapters.
- Sequencing: The final library is quantified and sequenced on an Illumina platform, typically generating 5-30 million single-end reads.
Protocol 3: Pyrosequencing for Methylation Quantification
- PCR of Bisulfite-Converted DNA: A target region (80-150 bp) is amplified from bisulfite-converted DNA using biotinylated primers designed specifically for bisulfite-converted sequence (avoiding CpG sites).
- Single-Stranded Template Preparation: The biotinylated PCR product is immobilized on streptavidin-coated sepharose beads. The beads are washed, and the non-biotinylated strand is denatured and removed with NaOH.
- Primer Annealing: A sequencing primer (adjacent to the CpG site(s) of interest) is annealed to the single-stranded template.
- Pyrosequencing Reaction: The template is sequentially incubated with DNA polymerase, ATP sulfurylase, luciferase, and apyrase, along with sequential dispensation of dNTPs (dATPS, dTTP, dGTP, dCTP). Incorporation of a nucleotide releases pyrophosphate, leading to a light signal proportional to the number of nucleotides incorporated.
- Data Analysis: The ratio of T (unmethylated) to C (methylated) signal at each interrogated CpG dinucleotide provides a precise percentage of methylation.
Visualized Workflows and Pathways
Title: WGBS and RRBS Library Preparation and Sequencing Workflow
Title: Pyrosequencing Quantitative Detection Principle
Title: CpG Methylation Leading to Transcriptional Silencing
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Bisulfite-Based Methylation Analysis
| Reagent / Kit | Primary Function | Critical Notes for Selection |
|---|---|---|
| DNA Bisulfite Conversion Kits (e.g., EZ DNA Methylation, Epitect, TrueMethyl) | Chemically converts unmethylated C to U with high efficiency and minimal DNA degradation. | Choose based on input DNA range (standard vs. low-input), automation compatibility, and desired elution volume. |
| Methylated & Unmethylated Control DNA | Positive and negative controls for bisulfite conversion, PCR, and sequencing assays. | Essential for validating the entire workflow and quantifying background noise. |
| Methylation-Specific PCR (MSP) Primers | Amplify bisulfite-converted DNA sequences specific to methylated or unmethylated alleles. | Requires meticulous design; use dedicated software (e.g., MethPrimer). |
| Pyrosequencing Assay Kits & Primers | Include pre-validated or custom-designed biotinylated PCR primers and sequencing primers for quantitative analysis. | Assays are target-specific; ensure primers avoid CpG sites and SNPs. |
| High-Fidelity, Bisulfite-Tolerant DNA Polymerase (e.g., Taq Gold, HotStarTaq, PyroMark PCR Master Mix) | PCR amplification of bisulfite-converted DNA, which is highly AT-rich and fragmented. | Must lack cytosine deamination activity (non-proofreading is common). |
| Methylated Adapters for NGS | Adapters for WGBS/RRBS library prep that are protected from bisulfite conversion, maintaining complementary sequences. | Critical for post-bisulfite amplification; contain methylated cytosines or specific base analogs. |
| Methylation Analysis Software (e.g., Bismark, BSMAP, PyroMark Q24, QUMA) | Aligns bisulfite-treated reads to a reference genome and calls methylation status at each cytosine. | Choice depends on method (WGBS/RRBS vs. Pyrosequencing) and computational resources. |
This technical guide compares two dominant platforms for genome-wide DNA methylation analysis—Illumina's EPIC microarrays and Next-Generation Sequencing (NGS)-based approaches—within the context of CpG island (CGI) methylation and gene silencing research. Understanding promoter CGI hypermethylation as a mechanism for transcriptional repression is fundamental in oncology, developmental biology, and therapeutic development. The choice of profiling technology significantly impacts the resolution, genomic coverage, and biological insights achievable in such studies.
Table 1: Core Technical Specifications and Performance Metrics
| Feature | Illumina Infinium MethylationEPIC (EPICv2.0) | NGS-Based Approaches (e.g., Whole-Genome Bisulfite Sequencing - WGBS; Targeted Panels) |
|---|---|---|
| Interrogated Cytosines | > 935,000 pre-defined CpG sites (EPICv2.0) | All ~28 million CpGs in human genome (WGBS) or custom selection (Targeted). |
| Genomic Coverage Focus | Predominantly CpG Islands, shores, shelves, enhancers, gene promoters. | Genome-agnostic (WGBS) or focused on regions of interest (Targeted). |
| Resolution | Single CpG resolution at defined sites. | Single-base-pair resolution across sequenced regions. |
| Sample Throughput | High (96+ samples per array run). | Low to Moderate (WGBS: ~12-24; Targeted: ~hundreds). |
| DNA Input Requirement | 250-500 ng (standard), 100 ng (low-input protocols). | WGBS: 100-200 ng (standard), <10 ng (ultra-low-input). Targeted: 10-50 ng. |
| Typical Sequencing Depth | Not Applicable (array intensity). | WGBS: 20-30x per strand; Targeted: 500-5000x. |
| Primary Data Analysis | IDAT files -> β/M-values (e.g., minfi, SeSAMe). | FASTQ -> Methylation calls (e.g., Bismark, BWA-meth, MethylDackel). |
| Approximate Cost per Sample (Reagents) | ~$150 - $300 | WGBS: ~$800 - $2000; Targeted: ~$100 - $500. |
| Key Advantage for CGI Research | Cost-effective, standardized for large cohorts; excellent coverage of known regulatory regions. | Discovery power; identifies novel/rare methylation events; absolute quantification. |
| Key Limitation | Discovery bias; cannot detect non-CpG methylation or novel loci. | Cost/complexity (WGBS); panel design required for targeted. |
Table 2: Suitability for Common Research Applications in Gene Silencing
| Research Application | Recommended Platform | Rationale |
|---|---|---|
| Biomarker Discovery & Validation (Large Cohorts) | EPIC Microarray | Lower cost and high throughput ideal for profiling hundreds of clinical samples. |
| Discovery of Novel Methylated Loci | WGBS or Enhanced EPIC* | Unbiased genome-wide coverage is essential for novel discovery. |
| High-Resolution Analysis of Specific Loci/Gene Panels | Targeted NGS (Bisulfite or Capture) | Extreme depth enables detection of low-frequency methylation in heterogeneous samples (e.g., tumors). |
| Integrative Multi-Omics (e.g., Methylation + Chromatin) | NGS-Based | Native compatibility with other NGS assays (ATAC-seq, ChIP-seq) for same-sample analysis. |
| Non-CpG (CHG/CHH) Methylation Studies | NGS-Based (WGBS) | EPIC does not probe non-CpG methylation. |
| Longitudinal / In-Vitro Drug Screening | EPIC or Targeted NGS | Balance of throughput, cost, and depth depending on scale and need for novel insights. |
Note: EPICv2.0 includes ~30% more coverage in enhancer regions compared to its predecessor, improving discovery capacity.
Detailed Experimental Protocols
Protocol: Standard Workflow for Illumina Infinium MethylationEPIC Array
Principle: Genomic DNA is bisulfite-converted, converting unmethylated cytosines to uracil (and later thymine), while methylated cytosines remain as cytosine. Converted DNA is amplified, fragmented, and hybridized to array beads. Single-base extension with fluorescently labeled nucleotides discriminates methylated (Cy5) from unmethylated (Cy3) alleles.
Detailed Steps:
- DNA Quantification & Quality Control: Use fluorometric assay (e.g., Qubit). Ensure integrity (RIN >7 via Bioanalyzer/TapeStation).
- Bisulfite Conversion: Process 250-500 ng genomic DNA using the EZ-96 DNA Methylation-Direct Kit (Zymo Research). Incubate at 98°C for 8 minutes, 64°C for 3.5 hours. Desulphonate and purify.
- Whole-Genome Amplification: Amplify converted DNA using the Infinium HD Assay Mega Kit. Incubate at 37°C for 20-24 hours.
- Enzymatic Fragmentation: Fragment amplified DNA enzymatically at 37°C for 1 hour.
- Precipitation & Resuspension: Isopropanol precipitate DNA, resuspend in hybridization buffer.
- Array Hybridization: Apply resuspended DNA to EPIC BeadChip. Hybridize in oven at 48°C for 16-24 hours with rocking.
- Single-Base Extension & Staining: Wash unhybridized DNA. Perform allele-specific single-base extension with labeled nucleotides (A/T labeled with Cy3; C/G labeled with Cy5). Stain BeadChip.
- Imaging: Scan BeadChip using the iScan or NextSeq 550 system. Generate IDAT intensity files.
Protocol: Whole-Genome Bisulfite Sequencing (WGBS) using Enzymatic Methylation Conversion
Principle: Genomic DNA is fragmented, and libraries are prepared. Bisulfite conversion is performed after adapter ligation (Post-Bisulfite Adapter Tagging, PBAT) to minimize DNA damage and input requirements. Sequencing provides single-base methylation status.
Detailed Steps (using Enzymatic Conversion):
- Library Preparation & Adapter Ligation: Fragment 10-100 ng genomic DNA via sonication (e.g., Covaris). End-repair, A-tail, and ligate methylated Illumina-compatible adapters.
- Bisulfite Conversion: Treat adapter-ligated DNA using the EM-Seq Kit (NEB). This two-enzyme process (TET2 and APOBEC) protects 5mC and 5hmC from deamination, converting unmodified C to U, thereby minimizing DNA degradation associated with traditional sodium bisulfite.
- PCR Amplification: Amplify libraries using a high-fidelity, uracil-tolerant polymerase (8-12 cycles). Index with barcodes for multiplexing.
- Library QC & Quantification: Assess library size distribution (Bioanalyzer). Quantify via qPCR (KAPA Library Quantification Kit).
- Sequencing: Pool libraries and sequence on an Illumina NovaSeq or HiSeq platform. Use 150 bp paired-end reads to maximize mapping efficiency. Aim for 20-30x physical coverage per strand.
- Bioinformatic Processing: Use a dedicated pipeline:
- Trimming & Quality Control: Trim adapters with Trim Galore! (with
--paired --clip_r1 15 --clip_r2 15 --three_prime_clip_r1 3 --three_prime_clip_r2 3 --max_n 1). - Alignment: Map to bisulfite-converted reference genome using
Bismark(Bowtie2 mode:bismark --genome <ref> -1 R1.fq -2 R2.fq --parallel 8). - Methylation Calling: Extract methylation calls:
bismark_methylation_extractor -p --gzip --bedGraph --parallel 8. Generate genome-wide coverage files.
- Trimming & Quality Control: Trim adapters with Trim Galore! (with
Platform Selection & Integration Pathways
Diagram Title: Decision Workflow for Methylation Platform Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for DNA Methylation Profiling Experiments
| Item | Function | Example Product (Supplier) |
|---|---|---|
| DNA Bisulfite Conversion Kit | Chemically converts unmethylated C to U, differentiating methylation states. Critical for both platforms. | EZ DNA Methylation Kit (Zymo Research), InnovaKits MethylEdge (Promega). |
| Methylation-Specific Array Kit | Contains all reagents for amplification, fragmentation, hybridization, staining, and scanning of EPIC arrays. | Infinium HD Assay Mega Kit (Illumina). |
| EM-Seq Kit | Enzymatic conversion alternative to bisulfite for NGS. Reduces DNA damage, improves library complexity. | NEBNext Enzymatic Methyl-seq Kit (New England Biolabs). |
| Methylated Adapter Kit | Provides adapters with methylated cytosines for WGBS/Targeted NGS to prevent digestion during bisulfite conversion. | TruSeq DNA Methylation Kit (Illumina), IDT for Illumina - UDI Adapters. |
| Uracil-Tolerant Polymerase | High-fidelity PCR enzyme capable of amplifying bisulfite-converted DNA (which contains uracil). | KAPA HiFi HotStart Uracil+ ReadyMix (Roche), Pfu Turbo Cx Hotstart (Agilent). |
| Methylation-Specific qPCR Controls | Validated controls for assay optimization, including fully methylated and unmethylated human genomic DNA. | Human Methylated & Non-methylated DNA Standard Set (Zymo Research). |
| Bisulfite Conversion Control Primers | PCR primers for unconverted and converted DNA to verify bisulfite conversion efficiency. | ACTB Conversion Control Primer Set (Zymo Research). |
| DNA Clean-up & Concentration Beads | For efficient purification and size selection of NGS libraries, especially post-bisulfite treatment. | AMPure XP Beads (Beckman Coulter). |
Data Analysis & Interpretation for CpG Island Silencing
Diagram Title: Core Data Analysis Pipeline for Methylation Studies
The EPIC microarray remains the workhorse for large-scale, cost-effective profiling of known regulatory elements, making it ideal for biomarker studies in CGI-mediated silencing. NGS-based approaches, particularly WGBS and targeted sequencing, offer unparalleled resolution and discovery potential for novel mechanisms. The emerging trend is towards integrated multi-omics, where NGS platforms provide a unified framework to correlate methylation with chromatin accessibility, histone modifications, and transcriptomics from the same sample. Furthermore, the development of long-read sequencing technologies (PacBio, Oxford Nanopore) promises to resolve haplotype-specific methylation and complex genomic contexts, representing the next frontier in understanding the complete epigenetic landscape of gene silencing.
Within the broader thesis context of CpG island (CGI) hypermethylation and its established role in transcriptional silencing of tumor suppressor genes, the need for precise, locus-specific analysis is paramount. While genome-wide methylation profiling identifies candidate loci, functional validation requires targeted assays. Methylation-Specific PCR (MSP) remains a cornerstone technique for this purpose, offering high sensitivity, specificity, and throughput for analyzing the methylation status of specific CpG dinucleotides within a region of interest, such as a promoter-associated CGI.
Core Principles of MSP
MSP relies on the bisulfite conversion of genomic DNA, which deaminates unmethylated cytosine to uracil (read as thymine during PCR), while methylated cytosine remains unchanged. Following conversion, two parallel PCR reactions are performed using primer sets specifically designed to amplify either the methylated (M) or unmethylated (U) converted sequence. The presence or absence of an amplicon in each reaction determines the methylation status of the target locus.
Assay Design: A Step-by-Step Protocol
1. Target Region Selection & In Silico Analysis
- Identify CGI: Using databases like UCSC Genome Browser, define the CGI overlapping your gene's promoter region of interest.
- Retrieve Sequence: Obtain ~500bp of genomic sequence spanning the CGI and transcription start site.
- In Silico Bisulfite Conversion: Use tools like MethPrimer or BiSearch to simulate bisulfite conversion.
- Input sequence:
CGCGATACGTCGATACGCG - Converted Methylated (M) strand:
CGCGATACGTCGATACGCG(C's remain) - Converted Unmethylated (U) strand:
UGUGATAUGUUGATAUGUGU(C's become U/T)
- Input sequence:
2. Primer Design Critical Parameters Primers must be specific to the bisulfite-converted sequence. Key design rules are summarized in Table 1.
Table 1: MSP Primer Design Parameters and Specifications
| Parameter | Methylated (M) Primer Set | Unmethylated (U) Primer Set | General Rule |
|---|---|---|---|
| CpG Site Placement | Must contain ≥1 CpG at the 3'-end. | Must contain NO CpG sites. Uses converted TpG sites. | 3'-specificity is critical for discrimination. |
| Length | 20-30 bp | 20-30 bp | - |
| Tm | 55-65°C | 55-65°C | Tm for M and U sets should be within 2°C. |
| Amplicon Size | 80-200 bp | 80-200 bp | Shorter products improve efficiency from degraded/converted DNA. |
| Sequence Validation | Must not bind to unconverted DNA or the U-converted sequence. | Must not bind to unconverted DNA or the M-converted sequence. | Use BLAST against bisulfite-converted genome. |
3. In Silico Validation & Specificity Check
- Perform in silico PCR against reference bisulfite-converted genomes (e.g., hg19/38 bisulfite-converted).
- Check for potential primer-dimer formation and secondary structure.
Experimental Protocol: Validating an MSP Assay
Materials Required: Genomic DNA & Bisulfite Conversion
- Purified Genomic DNA (100-500 ng, high integrity).
- Bisulfite Conversion Kit (e.g., EZ DNA Methylation-Lightning Kit, Qiagen Epitect).
- MSP Primer Sets (Validated M and U primers, resuspended in nuclease-free water).
- Hot-Start Taq Polymerase (Reduces non-specific amplification).
- PCR Reagents: dNTPs, MgCl₂, reaction buffer.
- Controls:
- Positive Methylated Control: DNA from a cell line with known methylation of the target (or in vitro methylated DNA).
- Positive Unmethylated Control: DNA from normal tissue or a cell line known to be unmethylated.
- No-Template Control (NTC): Water only.
- Agarose Gel Electrophoresis System (or capillary electrophoresis for quantitative analysis).
Workflow Protocol
- Bisulfite Conversion:
- Treat 200-500 ng of genomic DNA and controls according to kit protocol.
- Typical program: 98°C for 10 min, 64°C for 2.5 hours, hold at 4°C.
- Desulfonate and purify converted DNA. Elute in 20-40 µL. Store at -20°C.
MSP Reaction Setup:
- Prepare separate master mixes for M and U reactions.
- Per 25 µL reaction: 1X PCR buffer, 2.0-2.5 mM MgCl₂, 200 µM each dNTP, 0.4 µM each primer, 0.5-1.0 unit Hot-Start Taq, 2 µL bisulfite-converted DNA template.
- Cycling Conditions: Initial denaturation: 95°C for 5 min; 35-40 cycles of: 95°C for 30s, Primer-Specific Annealing Temp (Ta) for 30s, 72°C for 30s; Final extension: 72°C for 5 min.
Amplicon Detection & Analysis:
- Resolve 10 µL of each PCR product on a 2-3% agarose gel stained with ethidium bromide or SYBR Safe.
- Visualize under UV light. A valid result shows a band in the M reaction only for methylated DNA, U reaction only for unmethylated DNA, both for heterogeneously methylated samples, and no bands in NTCs.
The Scientist's Toolkit: Essential Reagent Solutions
Table 2: Key Research Reagents for MSP
| Reagent / Solution | Function in MSP | Critical Consideration |
|---|---|---|
| Sodium Bisulfite (Commercial Kits) | Chemically converts unmethylated C to U. | Conversion efficiency (>99%) is critical; optimized kits prevent DNA degradation. |
| Hot-Start Taq DNA Polymerase | Catalyzes PCR amplification. | Reduces non-specific priming and primer-dimer formation during setup, improving specificity. |
| DNA Methylation Standards (In vitro methylated & unmethylated human DNA) | Positive controls for M and U reactions. | Essential for assay validation and troubleshooting. |
| PCR Primers (Validated M/U sets) | Specifically anneal to bisulfite-converted methylated or unmethylated sequences. | 3'-end CpG placement (for M) and stringent in silico validation are non-negotiable. |
| Gel Visualization Dye (SYBR Safe) | Binds dsDNA for UV visualization. | Safer alternative to ethidium bromide; compatible with standard blue light transilluminators. |
Data Interpretation & Quantitative Considerations
While standard MSP is qualitative, it can be semi-quantified by gel densitometry. For true quantification, real-time MSP (qMSP) using fluorescent probes (e.g., TaqMan) is employed, providing a methylation ratio relative to a reference gene. Key performance metrics for a validated assay are summarized in Table 3.
Table 3: MSP Assay Validation Metrics and Typical Data
| Validation Metric | Target Performance | Example Experimental Result |
|---|---|---|
| Specificity | No amplification in incorrect channel (e.g., U primer set on fully methylated DNA). | M primers amplify only methylated control (Ct = 25). U primers show no amplification (Ct > 40). |
| Sensitivity | Detection of low-abundance methylated alleles in a background of unmethylated DNA. | Detectable amplification from a 1:1000 dilution of methylated DNA in unmethylated DNA. |
| Limit of Detection (LoD) | Minimum input DNA post-conversion required for reliable detection. | Robust amplification from 10 ng of bisulfite-converted DNA. |
| Reproducibility | Consistent Ct values or amplification patterns across replicates. | Intra-assay CV < 5% for qMSP Ct values. |
Visualization: MSP Workflow and Primer Design Logic
MSP Experimental Workflow from DNA to Result
Primer Design Logic for Methylated vs. Unmethylated Sequences
MSP is an indispensable tool for targeted validation of CGI methylation hypotheses generated from genome-wide studies. Rigorous in silico design, coupled with meticulous experimental validation using appropriate controls, ensures the generation of reliable, interpretable data on the methylation status of specific loci. When integrated with quantitative methods, MSP provides a powerful means to correlate epigenetic marks with gene silencing phenotypes, advancing our understanding in disease mechanisms and therapeutic targeting.
This whitepaper explores the pivotal role of single-cell methylomics in dissecting epigenetic heterogeneity, framed within the broader thesis of CpG island (CGI) methylation and its canonical function in transcriptional silencing. The inability of bulk assays to resolve cell-to-cell variation has obscured our understanding of epigenetic dynamics in development, tissue homeostasis, and tumor evolution. This guide details current methodologies, quantitative findings, and practical protocols, providing a technical resource for researchers and drug development professionals aiming to target the epigenetic landscape.
The established paradigm in epigenetics posits that hypermethylation of promoter-associated CpG islands leads to stable, heritable gene silencing, a hallmark of cancer (e.g., silencing of tumor suppressor genes). Bulk analyses, however, average methylation across thousands of cells, masking heterogeneous epigenetic states that drive phenotypic diversity. Single-cell methylomics transcends this limitation, enabling the mapping of epigenetic mosaicism within tissues and tumors. This resolution is critical for understanding clonal evolution, therapy resistance, and for identifying novel epigenetic biomarkers and drug targets.
Core Technological Platforms & Quantitative Comparisons
Current methodologies for single-cell DNA methylome profiling primarily involve bisulfite conversion (BS-conversion) followed by sequencing, with key variations in pre-amplification and library preparation.
Table 1: Comparison of Major Single-Cell Methylomics Methods
| Method | Core Principle | Approximate Genome Coverage (per cell) | Key Advantage | Primary Limitation |
|---|---|---|---|---|
| scBS-seq | Post-bisulfite tagging & amplification | 10-40% | High coverage uniformity; direct BS-conversion. | High sequencing cost; complex protocol. |
| sci-MET | Combinatorial indexing post-bisulfite | 1-5% | Extremely high throughput (1000s of cells). | Lower coverage per cell. |
| scWGBS (e.g., SMARTer) | Whole-genome amplification pre-BS | 5-20% | Robust commercial kits available. | Amplification bias; uneven coverage. |
| sn-m3C-seq (Multi-omic) | Simultaneous methylome & chromatin conformation | 5-15% (methylome) | Couples methylation with 3D genome structure. | Technically demanding; low throughput. |
Table 2: Representative Quantitative Findings from Recent Studies (2023-2024)
| Tissue/Tumor Type | Key Finding (Epigenetic Heterogeneity) | Measurement | Implication for CGI Silencing Thesis |
|---|---|---|---|
| Glioblastoma | 3-5 distinct epigenomic subclones per tumor | Variance in >50,000 CpGs | Subclones show differential hypermethylation of specific CGI promoters, correlating with expression of developmental genes. |
| Colorectal Adenoma | Intratumoral methylation entropy (disorder) predicts progression | Entropy score range: 0.15-0.85 | High entropy (mixed methylated/unmethylated cells at CGIs) indicates instability and higher malignant potential. |
| Healthy Hematopoiesis | ~2% of CpG sites show high cell-to-cell variance in progenitors | CV > 0.8 at variable sites | This "epigenetic noise" is enriched at lineage-specific CGI promoters, priming for cell fate decisions. |
| T-cell Exhaustion | Progressive CGI hypermethylation in exhausted vs. naive T-cells | Mean Δβ at key loci: +0.45 | Silencing of effector gene promoters via CGI methylation is a gradual, heterogeneous process in the tumor microenvironment. |
Detailed Experimental Protocols
Protocol: High-Throughput sci-MET for Sparse Methylome Profiling
This protocol is optimized for profiling thousands of single cells from a solid tumor digest.
I. Cell Preparation and Permeabilization
- Generate a single-cell/nuclei suspension from fresh-frozen tissue using a validated dissociation kit. Filter through a 40μm flow cytometry strainer.
- Critical: Perform nuclei isolation for archived tissues or tissues high in lipids. Use ice-cold lysis buffer (10mM Tris-HCl, 10mM NaCl, 3mM MgCl2, 0.1% NP-40, 1% BSA).
- Count and adjust concentration to ~1000 nuclei/μL. Use 0.2% formaldehyde for 5 min fixation, followed by quenching with 125mM glycine.
- Permeabilize nuclei with 0.2% Triton X-100 in PBS on ice for 15 min.
II. Combinatorial Indexing: Round 1 (96-Well Plate)
- Distribute ~100,000 permeabilized nuclei across a 96-well plate (~1000/well).
- In each well, perform in-well Tn5 tagmentation using a custom-loaded Tn5 transposase complex with well-specific indexed adapters (i7 index) in tagmentation buffer (10mM Tris, 5mM MgCl2, 10% DMF) at 55°C for 10 min. Quench with 0.1% SDS.
- Pool all wells. Nuclei are now tagged with a unique well-specific index.
III. Bisulfite Conversion and Nuclei Sorting
- Treat the pooled nuclei with sodium bisulfite using the EZ DNA Methylation-Lightning Kit (Zymo Research) following the manufacturer's protocol for large fragments. This deaminates unmethylated cytosines to uracils.
- Using fluorescence-activated nuclei sorting (FANS), sort nuclei into a 384-well plate containing lysis buffer, aiming for 1 nucleus per well based on DAPI signal. Confirm Poisson distribution for empty/ doublet rates.
IV. Combinatorial Indexing: Round 2 (384-Well Plate) & Library Prep
- In each 384-well plate, perform in-well PCR amplification (15-18 cycles) using a second set of indexed primers (i5 index). This step attaches the second combinatorial index and completes the adapter sequence.
- Pool all 384 wells. Clean the pooled library with solid-phase reversible immobilization (SPRI) beads.
- Perform a final bisulfite-PCR (8-10 cycles) to enrich for successfully converted fragments.
- Sequence on an Illumina NovaSeq platform with paired-end 150bp reads to achieve ~1-3x coverage per cell.
Protocol: Targeted Single-Cell CpG Island Methylation Profiling
For focused validation of CGI promoter silencing hypotheses.
I. Design and Synthesis of Padlock Probes
- Design ~100-200nt padlock probes targeting 50-100 CpG sites within your CGI(s) of interest. The probe ends should be complementary to the bisulfite-converted genomic sequence (C->T converted), with a 5' phosphate and a 3' adapter sequence.
- Include a unique molecular identifier (UMI) and a cell barcode region within the probe backbone.
II. Single-Cell Isolation and Bisulfite Conversion
- Isolate single cells via micromanipulation or fluidics into individual PCR tubes.
- Lyse cells and perform bisulfite conversion immediately using a single-tube protocol (e.g., Cells-to-CT kit adapted with bisulfite reagent).
- Neutralize and desalt the converted DNA.
III. Rolling Circle Amplification (RCA) and Sequencing
- Hybridize padlock probes to the converted single-cell DNA. Ligate using a Taq DNA ligase to circularize probes that find their exact target.
- Perform RCA using phi29 polymerase. This generates a long, single-stranded DNA concatemer containing many copies of the probe sequence.
- Fragment the RCA product and prepare sequencing libraries using a standard kit, amplifying with primers that capture the cell barcode and UMI.
- Sequence on a MiSeq (Illumina) for deep coverage of targeted sites. Align reads, accounting for C->T conversion, and extract methylation calls per CpG per cell using the UMI to correct for PCR duplicates.
Visualizations
Diagram 1: sci-MET Combinatorial Indexing Workflow (760px max-width)
Diagram 2: CGI Methylation to Gene Silencing Pathway (760px max-width)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Single-Cell Methylomics
| Item/Catalog | Function & Role in Protocol | Critical Notes |
|---|---|---|
| Chromium Next GEM Single Cell ATAC Kit (10x Genomics) | Adapted for nuclei isolation and tagmentation; provides a robust, microfluidics-based partitioning system. | Can be modified for post-capture bisulfite conversion workflows (scATAC-methyl). |
| EZ DNA Methylation-Lightning Kit (Zymo Research, D5030) | Rapid, efficient sodium bisulfite conversion of DNA in low-input and single-cell formats. | Essential for minimizing DNA degradation. Lightning kit is preferred for speed. |
| Tn5 Transposase (Illumina, 20034197) | Custom loading with adapter oligos allows for indexed tagmentation in combinatorial protocols. | Quality is critical for even tagmentation. Often loaded in-house for flexibility. |
| SMARTer Methyl-Seq Kit (Takara Bio, 634612) | Integrated kit for single-cell WGBS, using SMART amplification pre-bisulfite conversion. | Reduces protocol development time but may introduce amplification bias. |
| CellRaft AIR System (Cell Microsystems) | For precise, image-verified isolation of single cells into 96-well plates prior to targeted methylation assays. | Eliminates doublets and ensures single-cell origin for validation studies. |
| Phi29 DNA Polymerase (NEB, M0269S) | High-processivity enzyme for Rolling Circle Amplification (RCA) in targeted padlock probe assays. | Generates long, accurate copies for deep sequencing of target loci. |
| D1000 ScreenTapes (Agilent, 5067-5582) | For accurate size selection and quality control of libraries post-amplification and bisulfite conversion. | Critical for removing adapter dimers and optimizing sequencing efficiency. |
This whitepaper provides an in-depth technical guide for integrating DNA methylation data with transcriptomic profiles and chromatin state maps. Framed within a broader thesis on CpG island (CGI) methylation and gene silencing, this document addresses the mechanistic link between epigenetic marks, chromatin architecture, and gene expression outcomes. For researchers and drug development professionals, mastering this integrative approach is crucial for identifying novel therapeutic targets and biomarkers in complex diseases like cancer and neurological disorders.
Foundational Concepts & Current Landscape
DNA methylation at cytosine residues within CpG dinucleotides, particularly in promoter-associated CpG islands, is a canonical epigenetic mark associated with transcriptional repression. However, the relationship is not absolute; methylation in gene bodies can be associated with active transcription, and silencing can occur via mechanisms independent of promoter CGI methylation. Recent advances highlight the necessity of a multi-omics view: DNA methylation must be interpreted in the context of histone modifications (e.g., H3K4me3, H3K27me3), chromatin accessibility (ATAC-seq), and the resulting transcriptional output (RNA-seq).
Live search data (as of early 2025) confirms the trend towards single-cell multi-omics assays (e.g., scNMT-seq, scATAC-me) and spatial transcriptomics/methylomics, allowing correlation of epigenetic states with transcriptional activity within tissue architecture. Key challenges remain in data normalization, batch effect correction, and distinguishing correlation from causation.
Core Methodologies & Protocols
Experimental Protocols for Data Generation
Protocol 1: Whole-Genome Bisulfite Sequencing (WGBS) for Methylation Analysis
- Objective: Generate base-resolution DNA methylation maps.
- Procedure:
- DNA Extraction & Quality Control: Isolate high-molecular-weight DNA. Verify integrity via agarose gel electrophoresis or Bioanalyzer (RIN > 8.0).
- Bisulfite Conversion: Treat 100-500 ng DNA using a commercial kit (e.g., EZ DNA Methylation-Lightning Kit, Zymo Research). Incubate at 98°C for 10 minutes, 64°C for 2.5 hours. Desulphonate and purify.
- Library Preparation: Use a post-bisulfite adapter tagging method to minimize bias. Amplify with PCR (10-12 cycles).
- Sequencing: Perform paired-end 150 bp sequencing on an Illumina NovaSeq platform to a minimum depth of 20-30x coverage.
Protocol 2: RNA Sequencing (RNA-seq) for Transcriptomics
- Objective: Quantify gene expression levels and isoforms.
- Procedure:
- RNA Extraction: Use TRIzol or column-based kits with DNase I treatment. Assess quality (RIN > 9.0 for poly-A selection).
- Library Preparation: For mRNA-seq, perform poly-A selection. Use strand-specific library prep kits (e.g., NEBNext Ultra II). Fragment RNA to ~300 bp, synthesize cDNA, and add adapters.
- Sequencing: Sequence on an Illumina platform (75-100 million paired-end 75 bp reads per sample).
Protocol 3: Assay for Transposase-Accessible Chromatin with Sequencing (ATAC-seq)
- Objective: Map regions of open chromatin and nucleosome positions.
- Procedure:
- Nuclei Isolation: Lyse cells with cold lysis buffer, pellet nuclei (500 g, 10 min, 4°C).
- Tagmentation: Incubate 50,000 nuclei with Trb transposase and adapters (37°C, 30 min). Use a commercial kit (e.g., Illumina Tagment DNA TDE1 Kit).
- Purification & Amplification: Purify tagmented DNA. Amplify with PCR (5-10 cycles, determined by qPCR).
- Sequencing: Sequence paired-end on Illumina (50-100 million reads).
Computational Integration Workflow
The core analytical pipeline involves alignment, quantification, and joint analysis.
Diagram 1: Multi-Omics Data Integration Computational Workflow. (Max width: 760px)
Key Signaling Pathways in Epigenetic Silencing
The interplay between DNA methylation, histone modifications, and chromatin remodelers forms a reinforcing loop for stable gene silencing, often initiated at CpG island promoters.
Diagram 2: Pathway of CpG Island Methylation-Mediated Gene Silencing. (Max width: 760px)
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Application | Example Product/Catalog |
|---|---|---|
| Bisulfite Conversion Kit | Chemically converts unmethylated cytosines to uracil, leaving methylated cytosines intact for downstream sequencing or PCR. Critical for methylation analysis. | EZ DNA Methylation-Lightning Kit (Zymo Research, D5030) |
| Methylated & Unmethylated DNA Controls | Positive and negative controls for bisulfite conversion efficiency, PCR bias, and assay validation. | MilliporeSigma, D5014 & D5015 |
| DNMT/HDAC Inhibitors | Small molecule tools to perturb the epigenetic state (e.g., 5-Azacytidine for DNMT inhibition, Trichostatin A for HDAC inhibition). Used for functional validation. | Cayman Chemical, 10010212 & 89730 |
| Methylation-Specific PCR (MSP) Primers | For targeted validation of methylation status at specific loci post-genome-wide screening. | Designed using MethPrimer; synthesized by IDT. |
| ATAC-seq Kit | Optimized transposase and buffers for mapping open chromatin regions from low cell numbers. | Illumina Tagment DNA TDE1 Kit (20034197) |
| Methylated DNA IP (MeDIP) Kit | Antibody-based enrichment of methylated DNA fragments for reduced-representation methylation analysis. | Diagenode, mc-magme-003 |
| ChIP-seq Grade Antibodies | For mapping histone modifications (H3K4me3, H3K27me3, H3K9me3) to correlate with methylation states. | Active Motif, 39159 (H3K27me3) |
| Single-Cell Multi-Omics Kit | Enables simultaneous profiling of methylation and transcription from the same single cell. | 10x Genomics, Chromium Single Cell Multiome ATAC + Gene Expression |
Table 1: Expected Data Outputs from Core Multi-Omics Assays
| Assay | Typical Coverage/Depth | Key Output Metric | Common Software for Analysis | Data Format (Output) |
|---|---|---|---|---|
| WGBS | 20-30x genome-wide | Methylation β-value (0-1) per CpG | Bismark, MethylDackel, SeSAMe | bedGraph, bigWig |
| RRBS | 5-10x in CpG-rich regions | Methylation β-value per CpG | Bismark, BS-Seeker2 | bedGraph |
| RNA-seq | 20-40 million reads/sample (bulk) | TPM, FPKM, read counts | STAR, HISAT2, DESeq2, edgeR | .tsv, .csv |
| scRNA-seq | 50,000 reads/cell | UMI counts matrix | Cell Ranger, Seurat | mtx, h5ad |
| ATAC-seq | 50-100 million reads/sample | Insertion counts per base/peak | MACS2, ArchR (scATAC) | .bed, .narrowPeak |
Table 2: Correlation Strengths Between Promoter Methylation and Gene Expression
| Genomic Context | Typical Correlation with Expression | Interpretation & Notes |
|---|---|---|
| High-Density CpG Island (HCGI) Promoter | Strong Negative (ρ ≈ -0.7 to -0.9) | Methylation is strongly silencing, often involved in development & disease. |
| Low-Density CpG Island (LCGI) Promoter | Moderate Negative (ρ ≈ -0.4 to -0.6) | More variable effect; depends on tissue and transcription factor context. |
| Gene Body | Weak Positive (ρ ≈ 0.1 to 0.3) | Associated with active transcription, may prevent spurious initiation. |
| Enhancer Regions | Variable / Context-Dependent | Methylation often inversely correlates with enhancer activity (H3K27ac). |
| Intergenic Regions | Generally No Correlation | Most methylation is in repetitive elements, not directly regulating genes. |
Integrative multi-omics analysis moves beyond correlation to reveal the mechanistic hierarchy and feedback loops between DNA methylation, chromatin state, and transcription. For thesis research focused on CGI methylation and silencing, this approach is indispensable. It allows for the identification of bona fide epigenetically silenced driver genes versus passengers, and for the discovery of chromatin states that predispose to or result from methylation. As single-cell and spatial technologies mature, they will further refine these models, offering unprecedented resolution for drug discovery and personalized therapeutic strategies.
This whitepaper details the technical application of liquid biopsy for detecting CpG island (CGI) methylation in cell-free DNA (cfDNA) for cancer screening. It is framed within the broader, established thesis that aberrant hypermethylation of promoter-associated CpG islands is a primary mechanism of transcriptional silencing for tumor suppressor genes in carcinogenesis. The detection of these epigenetic alterations in cfDNA represents a non-invasive, sensitive, and specific modality for early cancer detection, minimal residual disease monitoring, and therapy response assessment.
Core Principles: CGI Methylation and cfDNA Biology
CpG Island Methylation & Gene Silencing: CpG islands are genomic regions with high frequency of CpG sites, typically found in gene promoters. In normal cells, these regions are usually unmethylated, permitting gene expression. The silencing thesis posits that hypermethylation of these CGIs, particularly in promoters of tumor suppressor genes (e.g., SEPT9, SHOX2, RASSF1A), recruits methyl-CpG-binding domain proteins and associated chromatin remodelers, leading to a transcriptionally repressive heterochromatin state. This is a fundamental and early event in many cancer pathways.
Cell-Free DNA in Cancer: Actively proliferating and dying tumor cells (via apoptosis, necrosis, and active secretion) release DNA fragments into the bloodstream. This circulating tumor DNA (ctDNA) carries the same genetic and epigenetic aberrations as the tumor of origin, including CGI hypermethylation signatures. cfDNA analysis involves the isolation and interrogation of this material from a standard blood draw.
Current Quantitative Landscape: Performance of Selected Methylation-Based Liquid Biopsy Assays
The following table summarizes recent data from key studies and commercially available assays focusing on CGI methylation detection in cfDNA for multi-cancer or specific cancer screening.
Table 1: Performance Metrics of Select CGI Methylation-Based Liquid Biopsy Assays
| Assay/Study (Cancer Type) | Target(s) | Sensitivity (Stage I-IV) | Specificity | Key Validation Cohort Size | Reference/Year |
|---|---|---|---|---|---|
| Guardant Health Shield (Colorectal Cancer Screening) | Multi-modal (incl. methylation of SEPT9 etc.) | 83% (for CRC) | 90% (in adults ≥45) | ~20,000 (ECLIPSE trial) | 2024 (Clinical Data) |
| GRAIL Galleri (Multi-Cancer Early Detection) | >1 million methylation sites (pan-cancer classifier) | 51.5% (across >50 cancers, Stage I-III) | 99.5% | ~6,600 (CCGA substudy) | 2021/2023 (Annals of Oncology) |
| ELSA-seq-based assay (Hepatocellular Carcinoma) | 519 CpG markers | 85.7% (Stage I HCC) | 94.3% | 1,091 (Training & Validation) | Nature Materials, 2023 |
| Plasma *SEPT9 Methylation (Epi proColon)* (Colorectal Cancer) | SEPT9 promoter methylation | 68.2% (all stages) | 79.0% | 7,941 (PROCOLON study) | Clin. Cancer Res., 2023 |
| Targeted Methylation Sequencing (Pancreatic Cancer) | 389 CpG sites | 67.3% (Stage I PDAC) | 96.0% | 853 (Case-Control) | Nature Comm., 2024 |
Detailed Experimental Protocols
Protocol: Targeted Bisulfite Sequencing for CGI Methylation Analysis in cfDNA
Objective: To quantitatively analyze methylation status at specific CpG islands in plasma-derived cfDNA.
Workflow Diagram Title: Targeted Bisulfite-seq for cfDNA Methylation
Materials & Reagents:
- Blood collection tubes (e.g., Streck Cell-Free DNA BCT, PAXgene Blood ccfDNA tubes).
- cfDNA extraction kit (e.g., QIAamp Circulating Nucleic Acid Kit, MagMAX Cell-Free DNA Isolation Kit).
- Bisulfite conversion kit (e.g., Zymo Research EZ DNA Methylation-Lightning Kit, Qiagen Epitect Fast DNA Bisulfite Kit).
- Targeted amplification primers or hybrid capture probes (e.g., Agilent SureSelectXT Methyl-Seq, Twist Bioscience NGS Methylation Panels).
- High-fidelity, bisulfite-converted DNA polymerase (e.g., KAPA HiFi HotStart Uracil+ ReadyMix).
- Library quantification kit (e.g., KAPA Library Quantification Kit for Illumina).
- Illumina sequencing platform (NextSeq 2000, NovaSeq X).
Procedure:
- Sample Collection & Processing: Collect 10-20 mL whole blood into stabilized tubes. Process within 6 hours: double centrifugation (e.g., 1600 x g for 10 min, then 16,000 x g for 10 min) to obtain platelet-poor plasma. Store at -80°C.
- cfDNA Extraction: Extract cfDNA from 3-5 mL plasma using a silica-membrane or bead-based kit according to manufacturer's protocol. Elute in 20-50 µL low-EDTA TE buffer. Quantify using a fluorescent dsDNA assay (e.g., Qubit dsDNA HS Assay).
- Bisulfite Conversion: Treat 10-50 ng cfDNA with sodium bisulfite using a commercial kit. This step converts unmethylated cytosines to uracils, while methylated cytosines (5mC) remain as cytosines. Purify and elute the converted DNA.
- Targeted Library Preparation:
- PCR-Based Approach: Perform multiplex PCR on bisulfite-converted DNA using target-specific primers designed for converted sequences. Use a limited cycle number (15-20 cycles). Clean up amplicons with SPRI beads.
- Hybrid Capture Approach: Construct a sequencing library from bisulfite-converted DNA with adaptor ligation. Perform hybrid capture using biotinylated RNA probes targeting regions of interest. Wash and elute the captured DNA.
- Library Amplification & Quantification: Amplify the final library (5-10 cycles). Purify with SPRI beads. Quantify library concentration via qPCR. Assess size distribution (e.g., Bioanalyzer).
- Sequencing: Pool libraries and sequence on an Illumina platform (2x150 bp recommended). Target a minimum mean coverage of 500-1000x per CpG site.
- Bioinformatic Analysis: Align reads to a bisulfite-converted reference genome (e.g., using Bismark or BWA-meth). Calculate methylation percentage at each CpG site as [Creads / (Creads + T_reads)] * 100. Apply machine learning classifiers trained on cancer/normal datasets to generate a cancer prediction score.
Protocol: Methylation-Specific Droplet Digital PCR (ddPCR) for Single-Gene Detection
Objective: Ultra-sensitive, absolute quantification of methylation at a specific CGI (e.g., SEPT9) in cfDNA.
Workflow Diagram Title: ddPCR for Methylation Detection
Materials & Reagents:
- Bisulfite-converted cfDNA (from Protocol 4.1, Step 3).
- ddPCR Supermix for Probes (No dUTP) (Bio-Rad).
- Methylation-specific (MS) and non-methylation-specific (U) primers and TaqMan probes (FAM-labeled for methylated, HEX/VIC-labeled for reference).
- DG8 cartridges, gaskets, and droplet generation oil (Bio-Rad).
- QX200 Droplet Generator and Reader (Bio-Rad).
- PCR plate heat sealer.
Procedure:
- Assay Design: Design primers/probes to specifically amplify the bisulfite-converted sequence of the methylated allele (CpG sites within the amplicon).
- Reaction Setup: Prepare a 20 µL ddPCR reaction mix containing 1x ddPCR Supermix, 900 nM primers, 250 nM probes, and ~5-10 ng of bisulfite-converted cfDNA.
- Droplet Generation: Load the reaction mix and 70 µL of droplet generation oil into a DG8 cartridge. Place in the QX200 Droplet Generator to create ~20,000 nanoliter-sized droplets per sample.
- PCR Amplification: Transfer the emulsified droplets to a 96-well PCR plate. Seal and run on a thermal cycler with optimized conditions (e.g., 95°C for 10 min, 40 cycles of 94°C for 30s and 60°C for 60s, 98°C for 10 min; ramp rate 2°C/s).
- Droplet Reading and Analysis: Place the plate in the QX200 Droplet Reader. The reader streams each droplet individually, measuring the fluorescence amplitude (FAM and HEX). Using QuantaSoft software, apply a fluorescence amplitude threshold to discriminate positive (methylated) from negative droplets. Calculate the absolute concentration (copies/µL) using Poisson statistics. Report as methylated genome equivalents per mL of plasma.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for CGI Methylation Analysis in cfDNA
| Item Category | Specific Example(s) | Function & Critical Notes |
|---|---|---|
| Blood Collection Stabilizers | Streck Cell-Free DNA BCT, PAXgene Blood ccfDNA tubes | Preserves blood cells to prevent genomic DNA contamination and cfDNA degradation during transport/storage. Critical for reproducible pre-analytics. |
| cfDNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit, MagMAX Cell-Free DNA Isolation Kit | High-efficiency, high-purity isolation of short-fragment cfDNA from large plasma volumes, minimizing inhibitor carryover. |
| Bisulfite Conversion Kits | Zymo Research EZ DNA Methylation-Lightning Kit, Qiagen Epitect Fast DNA Bisulfite Kit | Chemically converts unmethylated C to U with high efficiency while minimizing DNA degradation. Key determinant of final data quality. |
| Targeted Enrichment | Agilent SureSelect Methyl-Seq, Twist NGS Methylation Panels, MS-ddPCR primer/probe sets | Enables focused, deep sequencing of specific CGIs or genome-wide discovery. Design must account for bisulfite-converted sequence. |
| NGS Library Prep (Post-Bisulfite) | Accel-NGS Methyl-Seq DNA Library Kit, Swift Biosciences Accel-NGS Methyl-Seq | Optimized for the fragmented, single-stranded nature of bisulfite-converted DNA, improving complexity and yield. |
| Bioinformatics Tools | Bismark, BWA-meth, MethylKit, SeSAMe | Specialized aligners and analysis packages for handling bisulfite-converted reads, calling methylation states, and differential analysis. |
| Reference Standards | Horizon Discovery Methylation Multiplex cfDNA Reference Set | Commercially available multiplexed cfDNA with defined methylation patterns at specific loci. Essential for assay validation, QC, and inter-laboratory benchmarking. |
Navigating Experimental Pitfalls: Solutions for Reliable CGI Methylation Analysis
In the study of CpG island methylation and its role in gene silencing, bisulfite conversion remains the gold-standard technique for resolving 5-methylcytosine (5mC) from cytosine at single-nucleotide resolution. This chemical treatment deaminates unmethylated cytosines to uracil, while methylated cytosines remain unaffected. However, the harsh reaction conditions—low pH, high temperature, and prolonged incubation—inevitably lead to two critical artifacts: incomplete conversion (leading to false-positive methylation calls) and DNA degradation (resulting in loss of analyzable template and biased sequencing). For research focused on promoter CpG island hypermethylation as a mechanism of tumor suppressor gene silencing, these artifacts can severely confound data, leading to incorrect biological conclusions and hampering the identification of true epigenetic biomarkers for drug targeting.
The Artifacts: Mechanisms and Impacts
Incomplete Conversion
Incomplete conversion occurs when unmethylated cytosines fail to convert to uracil. This is often due to DNA secondary structure (e.g., hairpins in GC-rich CpG islands), insufficient bisulfite concentration, or suboptimal reaction time/temperature. The residual cytosine is subsequently read as "methylated" during PCR and sequencing, generating false-positive signals.
Quantitative Impact: Studies indicate that even low levels of incomplete conversion can significantly skew results. For example, in a sample with 0% true methylation, an incomplete conversion rate of 1% can lead to a reported methylation level of 1%, which is a critical error in low-methylation contexts.
DNA Degradation
The acidic and high-temperature conditions of bisulfite treatment catalyze the depurination and fragmentation of DNA. This results in:
- Reduced yields of long, amplifiable fragments.
- Biased amplification in downstream PCR, favoring shorter fragments and underrepresenting degraded regions.
- Complete loss of low-input samples (e.g., from biopsies or cell-free DNA).
Quantitative Impact: Standard bisulfite conversion can lead to >90% DNA loss, with fragment sizes often reduced to <300 bp. This is particularly detrimental for Next-Generation Sequencing (NGS) library preparation, which requires sufficient DNA integrity.
Table 1: Summary and Impact of Key Bisulfite Conversion Artifacts
| Artifact | Primary Cause | Consequence | Typical Quantitative Impact |
|---|---|---|---|
| Incomplete Conversion | DNA secondary structure, suboptimal reaction conditions | False-positive methylation calls | Can inflate reported methylation by 1-5% or more |
| DNA Degradation | Acidic pH, high temperature, long incubation | DNA fragmentation, loss of yield, PCR bias | >90% mass loss; fragment size <300 bp |
| PCR Bias | Degradation & sequence complexity post-conversion | Skewed representation of alleles/sequences | Can alter methylation frequency by >10% |
Detailed Experimental Protocols for Artifact Mitigation
Protocol: Quality Control Using Non-CpG Cytosine Conversion
To assess conversion efficiency, utilize cytosines in non-CpG contexts (e.g., CHH or CHG, where H = A, T, or C) as an internal control. In mammalian DNA, these sites are expected to be unmethylated in most somatic tissues.
- Spike-in Control: Include a synthetic, fully unmethylated DNA control (e.g., Lambda phage DNA) in every conversion batch.
- Post-Conversion Analysis: After sequencing, calculate the percentage of converted cytosines at all non-CpG sites.
- Threshold: Discard samples or batches with a conversion efficiency below 99.5%. Data analysis software (e.g., Bismark) typically provides this metric.
Protocol: Optimized Bisulfite Conversion for Fragile Samples
This protocol is adapted for low-input or highly fragmented DNA (e.g., from FFPE or plasma).
- Input DNA: 10-100 ng of DNA in a volume of ≤ 20 µL.
- Bisulfite Reagent: Use a commercial kit specifically formulated for low degradation (e.g., EZ DNA Methylation-Lightning Kit).
- Reaction Conditions:
- Denaturation: 98°C for 5 min (with kit-specific denaturation buffer).
- Incubation: 54°C for 60 minutes (with prepared bisulfite reagent). Note: Shorter, controlled incubation reduces degradation.
- Desalting: Bind DNA to provided spin-columns and desulfonate on-column.
- Elution: Elute in 10-20 µL of low-EDTA or EDTA-free TE buffer (pH 8.0). Store at -80°C if not used immediately.
- QC: Assess yield and fragment size using a high-sensitivity fluorometric assay (e.g., Qubit) and fragment analyzer.
Protocol: Post-Bisulfite Adapter Tagging (PBAT) for Highly Degraded DNA
PBAT minimizes the bias from DNA degradation by performing adapter ligation after bisulfite conversion, thereby protecting the converted strands.
- Perform bisulfite conversion on unligated genomic DNA (as in Protocol 3.2).
- First Strand Synthesis: Use a biotinylated primer complementary to a pre-ligated 3' adapter (or use random priming) to synthesize the first cDNA strand.
- Purification: Bind the biotinylated strand to streptavidin beads.
- Second Strand Synthesis: Synthesize the second strand, incorporating the full sequencing adapters.
- PCR Amplification: Perform a limited-cycle PCR to generate the final NGS library.
Visualizing Workflows and Relationships
Bisulfite Conversion Process and Artifact Introduction
Decision Workflow for Mitigating Bisulfite Artifacts
The Scientist's Toolkit: Essential Reagents & Materials
Table 2: Key Research Reagent Solutions for Overcoming Bisulfite Artifacts
| Item | Function & Rationale | Example Product/Type |
|---|---|---|
| Commercial Bisulfite Kits (Low-Degradation) | Formulations with optimized pH, stabilizing agents, and shorter protocols to maximize DNA integrity. | EZ DNA Methylation-Lightning Kit, MethylEdge Bisulfite Conversion System |
| High-Sensitivity DNA Assay Kits | Accurately quantifies low yields of fragmented DNA post-conversion. Critical for normalizing downstream PCR. | Qubit dsDNA HS Assay, TapeStation High Sensitivity D1000 |
| Unmethylated Control DNA | Serves as a spike-in control to experimentally determine the per-batch incomplete conversion rate. | Lambda Phage DNA, PCR-amplified non-mammalian DNA |
| Methylated Control DNA | Provides a positive control for conversion resistance of 5mC. | CpGenome Universal Methylated DNA |
| Post-Bisulfite Adapter Tagging (PBAT) Reagents | Specialized adapters and enzymes (e.g., BsmaI-resistant polymerases) for library prep from degraded DNA. | WGBS with PBAT kits, RBBS adapters |
| Bisulfite-Specific PCR Polymerases | Enzymes optimized for amplifying high-GC, converted templates with high fidelity and yield. | ZymoTaq Premix, EpiMark Hot Start Taq |
| Bisulfite Sequencing Data Analysis Software | Tools that calculate and optionally correct for incomplete conversion rates based on non-CpG cytosines. | Bismark, BSMAP, MethylKit (R package) |
The analysis of DNA methylation at CpG islands is foundational to understanding epigenetic regulation of gene silencing in development, disease, and therapeutic response. Bisulfite conversion remains the gold-standard technique, chemically deaminating unmethylated cytosines to uracils while leaving methylated cytosines intact. However, this process creates a non-homogeneous DNA template, introducing significant challenges for subsequent PCR amplification. Biased or non-specific amplification can lead to inaccurate quantification of methylation levels, directly compromising data integrity in studies linking hypermethylation of promoter-associated CpG islands to transcriptional silencing. This guide details a rigorous, evidence-based framework for primer design to ensure specific, unbiased amplification of bisulfite-converted DNA (bisDNA).
Core Principles and Challenges in Bisulfite Primer Design
The Post-Bisulfite Sequence Landscape
Bisulfite treatment creates a complex mixture of sequences from the original template. For a given genomic locus containing both methylated and unmethylated alleles, the resulting bisDNA has three potential sequence identities: original top strand, converted top strand, original bottom strand, and converted bottom strand. This effectively quadruples the sequence complexity in a heterogeneous sample.
- Amplification Bias: Preferential amplification of either methylated or unmethylated sequences due to primer mismatches.
- Strand Selection Bias: Inefficient priming for one of the four bisulfite strands.
- Primer Degeneracy Overload: Excessive degeneracy to accommodate C/T polymorphisms reduces primer specificity and annealing efficiency.
Strategic Primer Design Methodologies
Foundational Design Rules
The following table summarizes the critical, non-negotiable parameters for bisulfite-specific primers (BSPs).
Table 1: Core Parameters for Bisulfite-Specific Primer Design
| Parameter | Recommendation | Rationale |
|---|---|---|
| Length | 25-35 nucleotides | Ensures sufficient specificity despite reduced sequence complexity (A, T, G). |
| Tm | 57-62°C (ideal 60°C) | High, stringent Tm minimizes non-specific binding. Both primers should have Tm within 1°C. |
| CpG Sites | Avoid in primer 3' end. If unavoidable, use degenerate Y/R base. | A 3' mismatch at a CpG site causes severe amplification bias. |
| Non-CpG C's | Must be converted to Y (C/T) in the primer sequence. | Accounts for conversion of all unmethylated cytosines. |
| Product Size | 80-250 bp (optimal ≤150 bp) | BisDNA is fragmented; shorter products amplify more efficiently. |
| Specificity Check | In silico PCR against bisulfite-converted genome. | Verifies unique binding to the intended converted strand. |
Advanced Strategies to Eliminate Bias
Methylated vs. Unmethylated Allele-Specific Design:
- Methylation-Specific PCR (MSP): Uses two primer pairs. One pair matches the sequence where CpGs are methylated (C remains), the other matches where CpGs are unmethylated (converted to T). Requires prior bisulfite sequencing data.
- HeavyMethyl PCR: Employs a blocker oligonucleotide that binds to the unconverted (methylated) sequence, suppressing its amplification to enrich for unmethylated alleles, or vice-versa.
Bisulfite Sequencing Primer Design: For next-generation sequencing (NGS) applications, primers must include:
- Adapter Sequences: Illumina P5/P7 or other platform-specific adapters.
- Index/Barcode Sequences: For multiplexing.
- Target-Specific Sequence: Designed per Table 1 rules. A two-stage PCR (target-specific then adapter addition) is often preferred to minimize primer dimer formation.
Experimental Protocol for Primer Validation
Protocol: Sodium Bisulfite Conversion & PCR Optimization
Part A: Sodium Bisulfite Conversion (Using Commercial Kit)
- Input: 500 ng – 1 µg of high-quality genomic DNA in 20 µL elution buffer.
- Denaturation: Add 130 µL of CT Conversion Reagent (Kit Component). Incubate at 98°C for 10 minutes.
- Conversion: Incubate at 64°C for 2.5 hours in a thermal cycler with a heated lid (105°C).
- Binding: Load samples onto a spin column containing binding buffer.
- Desulfonation: Wash and apply desulphonation solution (0.2 M NaOH). Incubate at RT for 5-10 minutes.
- Wash & Elute: Perform multiple washes and elute in 20 µL of low-TE buffer or nuclease-free water. Store at -80°C.
Part B: PCR Setup & Touchdown Cycling
- Master Mix (25 µL reaction):
- 1X High-Fidelity PCR Buffer
- 200 µM each dNTP
- 0.4 µM each forward and reverse bisulfite primer
- 1 U of bisulfite-optimized hot-start DNA polymerase (e.g., ZymoTaq or similar)
- 2 µL of bisulfite-converted DNA template
- Touchdown Thermocycling Program:
- Initial Denaturation: 95°C for 5 min.
- 10 Cycles of Touchdown: Denature at 95°C for 30 sec, Anneal at 65°C (decreasing by 0.5°C per cycle) for 30 sec, Extend at 72°C for 45 sec.
- 35 Cycles of Standard Amplification: Denature at 95°C for 30 sec, Anneal at 60°C for 30 sec, Extend at 72°C for 45 sec.
- Final Extension: 72°C for 5 min.
- Hold at 4°C.
- Analysis: Run 5 µL of product on a 2% agarose gel. For quantitative analysis, use SYBR Green qPCR with melting curve analysis.
Table 2: Impact of Primer Design on Methylation Measurement Accuracy
| Design Flaw | % Bias in Methylation Quantification (qMSP) | Common Consequence |
|---|---|---|
| CpG at 3' end of primer | Up to 40% over/under-estimation | Allele-specific dropout, false positive/negative |
| Low Tm (<55°C) | Increased variability (SD >5%) | Non-specific amplification, high background |
| Excessive product length (>300 bp) | Reduced efficiency (E < 1.6) | Failed amplification from fragmented bisDNA |
| No in silico specificity check | Unquantifiable | Co-amplification of homologous sequences |
Visualizing the Workflow and Strategies
Bisulfite PCR Primer Design Strategy Map
The Scientist's Toolkit: Essential Reagent Solutions
Table 3: Key Research Reagents for Bisulfite PCR
| Reagent / Kit | Function in Workflow | Critical Feature for Bias Avoidance |
|---|---|---|
| DNA Bisulfite Conversion Kit (e.g., EZ DNA Methylation) | Chemical conversion of unmethylated C to U. | High conversion efficiency (>99.5%); minimal DNA degradation. |
| Bisulfite-Optimized DNA Polymerase (e.g., ZymoTaq, HotStarTaq Plus) | Amplification of bisulfite-converted templates. | Robust activity on bisDNA; hot-start to prevent mis-priming. |
| Methylated & Unmethylated Control DNA (e.g., CpGenome) | Positive controls for primer validation. | Universally methylated and in vitro methylated DNA. |
| PCR Purification Kit (e.g., MinElute) | Clean-up of bisulfite sequencing products. | High recovery of short, low-concentration amplicons. |
| Methylation-Agnostic qPCR Master Mix (e.g., SYBR Green) | Quantitative methylation analysis. | Uniform amplification efficiency for different sequence variants. |
| In Silico Design Tool (e.g., MethPrimer, BiSearch) | Primer design and specificity checking. | Algorithms optimized for bisulfite sequence degeneracy. |
Within CpG island methylation research, the fidelity of conclusions regarding gene silencing hinges on the accuracy of the primary methylation data. Meticulous primer design is not merely a preliminary step but a critical experimental variable. By adhering to stringent design rules, employing bias-minimizing strategies like MSP or HeavyMethyl, and rigorously validating primers with appropriate controls, researchers can ensure that their amplification step faithfully represents the true methylation state of the template. This vigilance safeguards against epigenetic artifacts, enabling robust correlation between promoter hypermethylation and transcriptional silencing in both basic research and clinical assay development.
Research into CpG island (CGI) methylation and gene silencing is foundational to understanding epigenetic regulation in development, disease, and therapeutics. A core thesis in this field posits that the aberrant hypermethylation of promoter-associated CGIs is a primary mechanism for the transcriptional silencing of tumor suppressor genes in cancer, representing a key target for epigenetic drug development. The validity of this thesis and the reliability of downstream data hinge on the precision of the measurement techniques used. This technical guide argues that the use of rigorously characterized, commercially sourced Fully Methylated and Unmethylated DNA Standards is not merely a best practice but a critical control that underpins all quantitative methylation analyses, ensuring accuracy, reproducibility, and meaningful biological interpretation.
The Role of Standards in Quantitative Methylation Analysis
Quantitative methods like bisulfite conversion followed by sequencing (Bisulfite-Seq) or pyrosequencing rely on the chemical deamination of unmethylated cytosines to uracils, while methylated cytosines remain unchanged. Inconsistencies in bisulfite conversion efficiency, PCR bias, and assay sensitivity can introduce significant error. Methylation standards serve as essential controls to:
- Calibrate the Assay: Establish a standard curve for absolute quantification.
- Monitor Conversion Efficiency: Unmethylated standards verify complete conversion (>99.5%).
- Assay Specificity: Methylated standards confirm the detection of methylated alleles.
- Determine the Limit of Detection/Quantification (LOD/LOQ): Essential for detecting low-level methylation in heterogeneous samples (e.g., liquid biopsies).
Key Data & Performance Metrics
The following table summarizes quantitative performance metrics validated using commercial DNA standards in typical methylation assays:
Table 1: Quantitative Performance Metrics Using Commercial Standards
| Metric | Definition | Target Value (Using Standards) | Impact of Non-Standardized Controls |
|---|---|---|---|
| Bisulfite Conversion Efficiency | % of unmethylated cytosines converted to uracil. | ≥99.5% | Inefficient conversion leads to false-positive methylation calls. |
| PCR Bias (Methylated vs. Unmethylated) | Ratio of amplification efficiency between alleles. | 1.0 (No bias) | Skewed amplification distorts true methylation ratios. |
| Assay Linearity (R²) | Correlation of expected vs. observed methylation % in standard mixtures. | >0.99 | Non-linearity invalidates quantitative results across the range. |
| Limit of Detection (LOD) | Lowest methylated allele fraction detectable. | Typically 0.1%-1% | Higher, unreliable LOD; inability to detect rare methylated events. |
| Inter-Assay Precision (CV) | Coefficient of variation for repeated measures of a standard. | <5% (for pyrosequencing) | High CV compromises longitudinal study data and treatment monitoring. |
Table 2: Common Commercial Sources & Specifications for DNA Standards
| Supplier | Product Example | Description | Key Application |
|---|---|---|---|
| Zymo Research | Human Methylated & Non-methylated DNA Set | Genomic DNA from a single source, treated in vitro with SssI methylase or sham-treated. | Gold standard for whole-genome assay calibration and bisulfite conversion control. |
| MilliporeSigma | CpGenome Universal Methylated DNA | Human DNA methylated in vitro with SssI methylase. | Used as a positive control for methylation-sensitive PCR and MSP. |
| Qiagen | EpiTect Control DNA | Pre-treated, ready-to-use bisulfite-converted DNA (methylated/unmethylated). | Control for post-bisulfite PCR and sequencing steps, removing conversion variability. |
Detailed Experimental Protocol: Using Standards for Pyrosequencing Assay Validation
Title: Protocol for Validating a Pyrosequencing Assay Using Methylation Standards
Objective: To establish a linear, sensitive, and precise pyrosequencing assay for a target CpG island.
Materials:
- Commercial Standards: Fully Methylated (100% M) and Unmethylated (0% M) Human Genomic DNA.
- Standard Mixtures: Serially diluted Methylated DNA in Unmethylated DNA (e.g., 100%, 75%, 50%, 25%, 10%, 5%, 0% M).
- Bisulfite Conversion Kit: (e.g., EZ DNA Methylation-Lightning Kit, Zymo Research).
- PCR Reagents: Primers designed for bisulfite-converted DNA (one biotinylated).
- Pyrosequencing System & Reagents: (e.g., Qiagen PyroMark Q48).
Methodology:
- Standard Mixture Preparation: Precisely mix the 100% M and 0% M standards to create the desired dilution series. Use a high-precision spectrophotometer or fluorometer for DNA quantification.
- Bisulfite Conversion: Convert 500 ng of each standard mixture and unknown samples in parallel using the same kit lot and thermal cycler. Include a "no-DNA" conversion control.
- PCR Amplification: Perform PCR on the bisulfite-converted DNA using optimized, bias-minimized primers. Amplify all standards and unknowns in the same run.
- Pyrosequencing: Process PCR products according to the Pyrosequencing workstation protocol. The dispensation order is defined by the sequence after bisulfite conversion.
- Data Analysis & Validation:
- Plot the observed methylation percentage (from the pyrosequencer software) for each standard mixture against its expected percentage.
- Calculate the linear regression (R²). A valid assay requires R² > 0.98.
- The 0% M standard confirms bisulfite conversion efficiency. Any signal >0.5% indicates incomplete conversion or background noise.
- The 100% M standard confirms assay specificity for the methylated allele.
- Use the standard curve to interpolate the methylation percentage of unknown samples.
- Calculate LOD/LOQ from replicate measures of the low-percentage standards (e.g., 5% and 1%).
Visualizing the Workflow and Logical Framework
Title: Methylation Analysis Workflow with Critical Control Points
Title: Logical Framework for Standards in Methylation Research
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Controlled Methylation Studies
| Reagent / Material | Function / Purpose | Critical Consideration |
|---|---|---|
| Fully Methylated DNA Standard | Positive control for methylated allele detection; calibrates the high end of the quantification range. | Must be in vitro methylated with SssI (CpG methylase) to ensure 100% CpG methylation. Human genomic background is ideal. |
| Unmethylated DNA Standard | Control for bisulfite conversion efficiency; calibrates the low end (0%) of quantification range. | Must be from a verified unmethylated source (e.g., in vitro amplified) or thoroughly treated to remove methylated DNA. |
| Bisulfite Conversion Kit | Chemically converts unmethylated C to U, while leaving 5mC unchanged. | Efficiency and DNA preservation vary. Kit must be validated with unmethylated standard every run. |
| Bias-Minimized PCR Primers | Amplify bisulfite-converted DNA without preferential amplification of methylated/unmethylated alleles. | Should be designed using specialized software, placed in CpG-free regions, and validated with standard mixtures. |
| Quantitative Methylation Platform | System for final readout (e.g., Pyrosequencer, MassARRAY, qPCR, NGS). | Platform-specific calibration using the same universal standards is required for cross-study comparison. |
Data Normalization and Batch Effect Correction in Genome-Wide Studies
In genome-wide studies of CpG island methylation, technical variability is an omnipresent confounder. Batch effects—systematic non-biological differences introduced during sample processing across different times, reagent lots, or personnel—can obscure true biological signals, such as the subtle methylation changes associated with gene silencing in cancer or development. Effective data normalization and batch correction are therefore not merely computational steps but foundational to deriving biologically valid conclusions. This technical guide details the core principles and state-of-the-art methodologies, framed within the critical context of DNA methylation research.
Batch effects arise from multiple stages of a typical genome-wide methylation study (e.g., Illumina EPIC array or bisulfite sequencing). Key sources include:
- Bisulfite Conversion Efficiency: Variability in conversion kits, incubation times, or temperature.
- Hybridization Conditions (for arrays): Lot-to-lot differences in array chips, hybridization ovens, or staining reagents.
- Sequencing Runs (for NGS): Differences in flow cells, sequencing chemistry versions, and cluster generation.
- DNA Quality & Quantity: Variation in input DNA integrity and concentration.
- Sample Processing Date: Drift in instrument calibration or ambient laboratory conditions.
Failure to address these effects can lead to false positives, false negatives, and irreproducible findings linking CpG island hypermethylation to promoter silencing.
Core Normalization Strategies
Normalization aims to remove technical variation within a batch to make samples comparable. The choice depends heavily on the assay platform.
Table 1: Common Normalization Methods for Methylation Data
| Method | Platform | Principle | Key Consideration for Methylation |
|---|---|---|---|
| Background Subtraction | Microarray | Subtracts nonspecific fluorescence signal. | Can be insufficient for severe dye bias. |
| Quantile Normalization | Microarray | Forces the empirical distribution of probe intensities to be identical across arrays. | Popular (e.g., in minfi), but may over-correct if large global biological differences exist. |
| Beta-Mixture Quantile (BMIQ) | Microarray | Separate normalization for Type I and Type II probes, which have different dynamic ranges. | Addresses a major platform-specific bias in Illumina arrays. |
| Subset Quantile Normalization (SWAN) | Microarray | Uses a subset of cross-reactive probes to guide normalization. | Performs well on Infinium 450k/EPIC arrays with diverse probe types. |
| Lambda Phage Spike-Ins | Bisulfite Seq | Uses unmethylated spike-in controls to estimate and correct for conversion efficiency. | Requires experimental forethought; excellent for absolute methylation estimation. |
Batch Effect Correction Algorithms
Once normalized, data must be adjusted to remove variation between batches. These methods model and subtract the batch-associated component.
Table 2: Batch Effect Correction Algorithms
| Algorithm | Model Type | Key Feature | Suitability for Methylation |
|---|---|---|---|
| ComBat (Empirical Bayes) | Linear Model | Estimates batch-specific parameters (location, scale) and shrinks them toward the global mean. Robust to small batch sizes. | Widely used; effective when batch is known and biological phenotype is not confounded with batch. |
| Remove Unwanted Variation (RUV) | Factor Analysis | Uses control probes/genes (e.g., negative control probes) or replicates to estimate unwanted factors. | Ideal when technical factors are unknown or complex. Requires careful selection of controls. |
| Surrogate Variable Analysis (SVA) | Factor Analysis | Identifies latent factors of variation, both biological and technical, without prior knowledge of batch. | Powerful for unknown confounders, but risk of removing biological signal. |
| Harmony | Iterative PCA | Clusters cells/samples in PCA space and corrects them to be aligned across batches. | Originally for single-cell, but applicable to bulk data; effective for large, complex batch structures. |
| limma (removeBatchEffect) | Linear Model | Fits a linear model to the data and removes components attributable to batch. | Straightforward and effective when design is simple and batch is known. |
An Integrated Experimental & Computational Workflow
A robust analysis pipeline integrates both wet-lab and computational best practices.
Experimental Protocol: Minimizing Batch Effects in a Methylation Study
Study Design:
- Randomization: Process cases and controls across all batches randomly.
- Balancing: Ensure key biological groups (e.g., tumor/normal) are equally represented in each batch.
- Replication: Include at least one technical replicate (same sample) in a different batch to assess batch effect magnitude.
Wet-Lab Processing:
- Use a single, consistent protocol for bisulfite conversion (e.g., Zymo EZ DNA Methylation Kit).
- For arrays, process all samples using the same chip lot and reagent kit lot if possible.
- For sequencing, use unique dual indexes (UDIs) to prevent index hopping and pool samples across conditions for each library preparation batch.
- Include control samples: known methylation standards (fully methylated/unmethylated DNA) and negative controls.
Computational Pipeline:
- Quality Control (QC): Assess bisulfite conversion efficiency, signal intensities, and detect outliers.
- Normalization: Apply platform-specific normalization (e.g., BMIQ for arrays, bias correction for seq).
- Batch Detection: Perform PCA or MDS plotting, color samples by processing date/lab. Statistically test for batch association (PERMANOVA).
- Correction: Apply a chosen batch correction method (e.g., ComBat), ensuring the model does not include the biological variable of interest.
- Validation: Verify correction by visualizing PCA plots post-adjustment. Ensure biological replicates cluster. Confirm known positive controls (e.g., imprinted genes) remain significant.
Diagram: Integrated Workflow for Methylation Data Analysis
Table 3: Research Reagent Solutions for Methylation Studies
| Item | Function & Relevance | Example Product/Brand |
|---|---|---|
| Bisulfite Conversion Kit | Chemically converts unmethylated cytosines to uracils, while methylated cytosines remain unchanged. The critical first step. | Zymo Research EZ DNA Methylation Kit, Qiagen EpiTect Fast. |
| DNA Methylation Standards | Fully methylated and fully unmethylated genomic DNA controls. Used to assess conversion efficiency and as assay positive controls. | MilliporeSigma CpGenome Universal Methylated DNA, Zymo Human HCT116 DKO Methylated DNA. |
| Infinium Methylation Array | Genome-wide bead-chip platform for profiling methylation at known CpG sites. Standard for large cohort studies. | Illumina Infinium MethylationEPIC v2.0 BeadChip. |
| Methylated DNA Spike-in Controls | Synthetic DNA with known methylation patterns added to samples pre-conversion. Allows absolute quantification in sequencing. | Cambridge Epigenetix SNAP-Cell spike-in mixes. |
| High-Fidelity PCR Master Mix for Bisulfite DNA | Amplifies bisulfite-converted, uracil-containing DNA with high efficiency and minimal bias. Critical for sequencing library prep. | NEB Next Ultra II Q5 Master Mix, Qiagen PyroMark PCR Kit. |
| Methylation-Sensitive Restriction Enzymes (MSRE) | Enzymes that cut only unmethylated recognition sites. Used in validation (qPCR) or targeted approaches. | New England Biolabs (e.g., HpaII). |
| Bioinformatics Software | Packages for normalization, batch correction, and differential analysis. | R/Bioconductor: minfi, sva, ChAMP, DSS. Python: MethylSig, pyComBat. |
Validation: The Crucial Final Step
After correction, validation is mandatory:
- Visual Inspection: PCA/MDS plots should show batches intermingled, while biological groups separate.
- Statistical Confirmation: The association between principal components and batch should be minimized (high p-value in PERMANOVA).
- Biological Verification: Known differentially methylated regions (DMRs) associated with gene silencing (e.g., hypermethylated promoters of tumor suppressor genes like CDKN2A or MLH1) should be recoverable and validated by an orthogonal method (e.g., pyrosequencing or Methylation-Specific PCR (MSP)).
Diagram: Post-Correction Validation Logic Flow
In the study of CpG island methylation and its functional consequence in gene silencing, rigorous data normalization and batch effect correction are non-negotiable components of the analytical pipeline. By integrating thoughtful experimental design with a structured computational workflow—encompassing platform-specific normalization, careful application of correction algorithms like ComBat or SVA, and thorough validation—researchers can ensure that the observed methylation differences truly reflect underlying biology. This discipline is fundamental for discovering robust epigenetic biomarkers and understanding mechanisms of disease.
Within the broader thesis on CpG island methylation and gene silencing research, establishing causality remains a paramount challenge. Observational studies frequently reveal correlations between hypermethylation of promoter-associated CpG islands and gene silencing. However, these associations do not prove that methylation is the causative agent of silencing, as it could be a consequence or a parallel epigenetic mark. This technical guide details the application of targeted epigenome editing tools, specifically CRISPR-dCas9 fused to the catalytic domains of TET1 (Ten-Eleven Translocation 1) or DNMT3A (DNA Methyltransferase 3A), to functionally validate the causal role of DNA methylation in gene regulation.
Core Principles of Targeted Epigenome Editing for Validation
The CRISPR-dCas9 system provides a programmable DNA-targeting platform. By fusing dCas9 (nuclease-dead Cas9) to epigenetic effector domains, researchers can directly manipulate the epigenetic state at a specific genomic locus without altering the underlying DNA sequence.
- CRISPR-dCas9-DNMT3A: Targets and induces de novo DNA methylation at CpG sites within a specified genomic window, typically a promoter region. This tests whether adding methylation is sufficient to cause silencing.
- CRISPR-dCas9-TET1: Targets and catalyzes the oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and further derivatives, initiating active DNA demethylation. This tests whether removing methylation is sufficient to reactivate a silenced gene.
The reversal of a phenotype (silencing or activation) upon targeted intervention provides robust evidence for causation.
Table 1: Representative Quantitative Outcomes from Functional Validation Studies
| Target Gene & Context | Intervention (Effector) | Methylation Change at Target (%) | Gene Expression Change (Fold) | Key Measured Phenotype | Primary Reference (Example) |
|---|---|---|---|---|---|
| TIMP3 (Tumor Suppressor) in HeLa Cells | dCas9-DNMT3A | +35 to +50 (CpG island) | -8 to -12 (mRNA) | Increased Cell Invasion | Liu et al., 2016 |
| BRCA1 (Tumor Suppressor) in MCF-7 Cells | dCas9-DNMT3A | +40 | -15 (mRNA) | Reduced DNA Repair Capacity | McDonald et al., 2016 |
| MGMT (Silenced in Glioma) in U87 Cells | dCas9-TET1 | -60 (promoter) | +25 (mRNA) | Sensitization to Temozolomide | Huang et al., 2017 |
| FMR1 (in FXS iPSCs) | dCas9-TET1 | -30 (CGG expansion) | +5 to +8 (mRNA) | Partial Reactivation | Liu et al., 2018 |
| IL6ST Promoter in Primary T Cells | dCas9-DNMT3A | +25 | -20 (protein) | Attenuated STAT3 Signaling | Lei et al., 2017 |
Detailed Experimental Protocol
Protocol: Validating a Putative Silenced Tumor Suppressor Gene via CRISPR-dCas9-DNMT3A-Mediated Methylation
Objective: To determine if targeted promoter hypermethylation is sufficient to silence a gene whose expression is correlated with, but not proven to be caused by, low methylation in cell lines.
Materials:
- Cell Line: A cell line where the gene of interest (GOI) is expressed and its promoter is in a hypomethylated state.
- Plasmids: pLV-dCas9-DNMT3A (or DNMT3A catalytic domain: DNMT3ACD) and pLV-sgRNA expression vectors. A non-targeting sgRNA (NT-sgRNA) control is essential.
- Reagents: Lentiviral packaging plasmids (psPAX2, pMD2.G), transfection reagent (e.g., PEI), polybrene, puromycin.
Procedure: Week 1: Design and Cloning
- Design 3-5 sgRNAs targeting the CpG island within the promoter region (approx. -500 to +500 bp from TSS) of the GOI. Use tools like CHOPCHOP or CRISPick.
- Clone sgRNA sequences into the pLV-sgRNA backbone via BsmBI restriction site ligation or Gibson assembly. Sequence-verify clones.
Week 2-3: Lentivirus Production and Transduction
- Co-transfect HEK293T cells with the packaging plasmids (psPAX2, pMD2.G) and either pLV-dCas9-DNMT3A or pLV-sgRNA plasmids using PEI.
- Harvest lentiviral supernatant at 48 and 72 hours post-transfection.
- Transduce target cells sequentially: first with dCas9-DNMT3A virus, select with puromycin (e.g., 2 µg/mL) for 5 days. Then transduce stable cells with pooled sgRNA viruses, select with a second antibiotic (e.g., blasticidin).
Week 4: Validation and Analysis
- Genomic DNA Extraction: Harvest cells from experimental (GOI-sgRNA) and control (NT-sgRNA) groups.
- Bisulfite Sequencing (Bisulfite Pyrosequencing or NGS): Treat DNA with sodium bisulfite, PCR amplify the targeted promoter region, and quantify CpG methylation percentage. This confirms the epigenetic manipulation.
- RNA Extraction and qRT-PCR: Isolate total RNA, synthesize cDNA, and perform qPCR for the GOI and housekeeping genes (e.g., GAPDH, ACTB). Calculate relative expression (ΔΔCt method).
- Phenotypic Assay: Perform a relevant functional assay (e.g., proliferation assay, apoptosis assay, invasion/migration assay) to link epigenetic silencing to cellular phenotype.
- Off-target Analysis: Perform reduced representation bisulfite sequencing (RRBS) or targeted bisulfite sequencing of predicted off-target sites to assess specificity.
Protocol: Reactivation via CRISPR-dCas9-TET1-Mediated Demethylation
The protocol mirrors 4.1, with key substitutions:
- Cell Line: Use a cell line where the GOI is endogenously silenced and promoter-hypermethylated.
- Plasmid: pLV-dCas9-TET1CD (catalytic domain).
- Analysis: Confirm loss of 5mC and gain of 5hmC at the target via hydroxymethylated DNA immunoprecipitation (hMeDIP) or oxidative bisulfite sequencing (oxBS-seq). Measure gene reactivation via qRT-PCR and protein analysis (western blot).
Visualization of Core Concepts and Workflows
Title: Logic Flow from Correlation to Causal Validation
Title: Mechanism of Targeted Epigenetic Editing for Validation
Title: Step-by-Step Validation Experiment Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Functional Validation Studies
| Reagent / Material | Function in Experiment | Key Considerations / Examples |
|---|---|---|
| dCas9-Effector Plasmids | Core constructs expressing nuclease-dead Cas9 fused to epigenetic writer/eraser. | dCas9-DNMT3A (or DNMT3ACD): From Addgene (#113158). dCas9-TET1CD: From Addgene (#113156). Use catalytically inactive mutants as controls. |
| sgRNA Cloning Backbone | Vector for expression of single guide RNA targeting the genomic locus of interest. | Commonly used: pLV-sgRNA (lentiviral), pXPR_XXX (Addgene). Contains BsmBI sites for cloning. |
| Lentiviral Packaging Plasmids | For production of safe, integration-competent viral particles to deliver constructs to cells. | 2nd/3rd Generation: psPAX2 (gag/pol), pMD2.G (VSV-G envelope). Essential for hard-to-transfect cells (e.g., primary cells). |
| Bisulfite Conversion Kit | Chemically converts unmethylated cytosines to uracil, leaving 5mC unchanged, enabling methylation analysis. | Gold Standard: EZ DNA Methylation kits (Zymo Research) or EpiTect Bisulfite kits (Qiagen). Efficiency >99% is critical. |
| Pyrosequencing System | Quantitative, real-time sequencing of bisulfite-converted DNA to measure methylation percentage at individual CpGs. | Platform: PyroMark Q96 (Qiagen). Software: PyroMark CpG Assay Design for primer design. High accuracy for low-plex targets. |
| Antibodies for 5mC/5hmC | Immunochemical detection of global or locus-specific methylation/hydroxymethylation changes. | 5mC: Clone 33D3. 5hmC: Clone 4D9. Used for dot-blot, immunofluorescence, or hMeDIP-seq. |
| Next-Generation Sequencing Service | For comprehensive on-/off-target analysis (e.g., whole-genome bisulfite sequencing, RRBS). | Provides unbiased assessment of editing specificity. Critical for rigorous validation studies. |
| Positive Control Cell Line | A cell line with a well-characterized, methylation-silenced gene (e.g., MGMT in glioblastoma). | Serves as a benchmark for demethylation/reactivation efficiency of the dCas9-TET1 system. |
Best Practices for Sample Collection, Storage, and DNA Isolation to Preserve Methylation Status
Within the context of advancing research on CpG island methylation and its role in transcriptional gene silencing, the integrity of epigenetic analyses is fundamentally dependent on pre-analytical variables. This guide details rigorous protocols for sample handling to ensure the preservation of DNA methylation patterns, a critical factor for biomarker discovery and epigenetic drug development.
Sample Collection & Immediate Handling
The initial steps are paramount to prevent methylation drift. Different sample types require tailored approaches.
Blood and Bone Marrow
For peripheral blood mononuclear cells (PBMCs) or circulating cell-free DNA (ccfDNA):
- Collection Tubes: Use EDTA or specialized cell-stabilizing tubes (e.g., PAXgene Blood DNA tubes) to prevent in vitro leukocyte activation and enzymatic degradation.
- Processing Time: Isolate PBMCs or plasma within 2 hours at room temperature (RT) or within 24 hours if stored at 4°C. For ccfDNA methylation studies, plasma separation should occur within 2 hours to minimize leukocyte lysis contamination.
- Protocol: PBMC Isolation from EDTA Blood
- Dilute blood 1:1 with PBS.
- Layer over Ficoll-Paque PLUS density gradient medium.
- Centrifuge at 400 × g for 30 minutes at RT, with brake off.
- Harvest the PBMC interface layer.
- Wash twice with PBS.
- Pellet cells for immediate DNA extraction or stabilization.
Tissue Biopsies
Solid tissues are highly susceptible to ischemia.
- Ischemia Time: Minimize time to preservation to <30 minutes. Document warm and cold ischemia times meticulously.
- Preservation Methods:
- Flash-Freezing: Optimal. Snap-freeze in liquid nitrogen within minutes of excision. Store at -80°C.
- Stabilization Solutions: Commercial nucleic acid stabilizers (e.g., RNAlater) can preserve methylation at 4°C for 24 hours before long-term storage at -80°C. Note: Formalin-fixed, paraffin-embedded (FFPE) tissue is suboptimal due to DNA fragmentation and potential cytosine deamination, but specialized protocols exist.
Cell Cultures
- Harvesting: Use gentle trypsinization and immediate neutralization. Avoid prolonged enzymatic treatment.
- Washing: Wash pellets twice with cold PBS.
Sample Storage
Long-term storage conditions must halt all enzymatic activity.
Table 1: Recommended Storage Conditions by Sample Type
| Sample Type | Short-term (≤1 week) | Long-term (>1 week) | Critical Consideration |
|---|---|---|---|
| Cell Pellet | -20°C | -80°C (preferred) or liquid N₂ | Avoid repeated freeze-thaw cycles. |
| Tissue | Not recommended | -80°C (flash-frozen) or liquid N₂ | Store in small aliquots. |
| DNA (Isolated) | 4°C (in TE buffer, pH 8.0) | -20°C or -80°C (for >1 year) | TE buffer prevents acid hydrolysis. |
| Plasma/Serum | 4°C | -80°C | Single freeze-thaw cycle acceptable for ccfDNA. |
DNA Isolation: Methodology and Pitfalls
The choice of isolation method directly impacts DNA quality, fragment size, and methylation fidelity.
Table 2: Comparison of DNA Isolation Methods for Methylation Analysis
| Method | Principle | Bisulfite Conversion Yield | Risk of Methylation Loss | Best For |
|---|---|---|---|---|
| Phenol-Chloroform | Organic extraction, ethanol precipitation. | Variable; can be lower. | High. Harsh pH and prolonged processing can demethylate. | Legacy protocols; not recommended for de novo studies. |
| Silica-Column (Most Kits) | Binding in high chaotropic salt, wash, elute. | High (with optimized kits). | Low-Moderate. Ensure lysis is performed at neutral pH. | High-quality DNA from fresh/frozen samples. |
| Magnetic Beads | Paramagnetic bead binding in PEG/salt buffer. | High. | Very Low. Rapid, gentle, and automatable. | High-throughput studies, ccfDNA, PBMCs. |
| Salting-Out | Protein precipitation with saturated NaCl. | Moderate. | Moderate. Simpler than phenol but less pure. | Large-scale genomic DNA from blood. |
Critical Protocol: Methylation-Preserving DNA Extraction (Magnetic Bead-Based)
Reagents: Lysis buffer (Proteinase K, Tris-HCl pH 8.0, EDTA, SDS), binding buffer (PEG, NaCl), wash buffers (ethanol-based), elution buffer (10 mM Tris-HCl, pH 8.5), magnetic beads.
- Lysis: Resuspend cell/tissue sample in lysis buffer. Incubate at 56°C for 1-2 hours with agitation. Keep pH neutral.
- Binding: Add binding buffer and magnetic beads to lysate. Incubate at RT for 5 minutes.
- Capture: Place tube on a magnetic rack. Discard supernatant once clear.
- Washing: Wash beads twice with 80% ethanol while on the magnet. Dry briefly (~5 min).
- Elution: Resuspend beads in elution buffer. Incubate at 55°C for 5 minutes. Capture beads and transfer purified DNA supernatant to a new tube.
- QC: Quantify by fluorometry (e.g., Qubit). Assess integrity via agarose gel or Fragment Analyzer.
Pre-Bisulfite Conversion Quality Control
Before proceeding to bisulfite sequencing or array analysis, assess DNA quality with metrics relevant to epigenetic assays.
Table 3: Pre-Analytical Quality Control Metrics
| Metric | Target | Method | Rationale |
|---|---|---|---|
| DNA Concentration | >10 ng/µL for most assays. | Fluorometric assay (Qubit). | More accurate than A260 for dilute samples. |
| Purity (A260/280) | 1.8 - 2.0. | Spectrophotometry (NanoDrop). | Indicates protein/phenol contamination. |
| Integrity (DV200) | >70% for FFPE; >50% for ccfDNA. | Fragment Analyzer, TapeStation. | % of fragments >200 bp, critical for library prep. |
| Degradation | Clear high-molecular-weight band. | Agarose gel electrophoresis. | Visual check for smearing. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Methylation-Preserving Workflows
| Item | Function | Example |
|---|---|---|
| PAXgene Blood DNA Tube | Stabilizes blood cells at collection, preventing gene expression changes and DNA degradation. | QIAGEN PAXgene Blood DNA Tube |
| RNAlater Stabilization Solution | Preserves nucleic acids in tissues at 4°C, allowing delayed freezing without degradation. | Thermo Fisher Scientific RNAlater |
| Proteinase K | Digests nucleases and other proteins during cell lysis, protecting DNA. | Roche Proteinase K |
| Magnetic Bead DNA Purification Kit | Enables rapid, gentle, high-yield DNA isolation with minimal methylation damage. | MagMAX DNA Multi-Sample Kit |
| Bisulfite Conversion Kit | Converts unmethylated cytosines to uracil while leaving 5-methylcytosine intact. | EZ DNA Methylation-Lightning Kit |
| Fluorometric DNA QC Assay | Accurately quantifies double-stranded DNA without interference from RNA or contaminants. | Invitrogen Qubit dsDNA HS Assay |
| Methylation-Specific PCR (MSP) Primers | Amplify sequences based on methylation status post-bisulfite conversion. | Custom-designed primers |
| Methylation Array | Genome-wide profiling of methylation states at CpG sites. | Illumina Infinium MethylationEPIC BeadChip |
Workflow and Pathway Visualizations
Diagram 1: Sample Integrity Workflow for Methylation Analysis
Diagram 2: CpG Methylation Leads to Transcriptional Silencing
Benchmarking Insights: Validating CGI Methylation Patterns Across Tissues, Diseases, and Model Systems
Within the broader thesis on CpG island (CGI) methylation and gene silencing research, the central challenge is distinguishing epigenetic noise from pathogenic signal. Tissue-specific methylomes represent the foundational reference maps, cataloging the normal, programmed variation in DNA methylation across cell types. This guide details the technical frameworks for defining these baselines and identifying disease-associated alterations that disrupt transcriptional programs, with a focus on CGI hypermethylation and promoter silencing in oncogenesis and complex disorders.
Core Concepts: Normal Tissue-Specific Methylation
Normal variation is established during differentiation, creating stable epigenetic landscapes that define cellular identity. Key features include:
- Tissue-Specific Differentially Methylated Regions (tsDMRs): Methylation patterns unique to a cell lineage.
- Partially Methylated Domains (PMDs) vs. Highly Methylated Domains (HMDs): Broad genomic structures with distinct methylation levels.
- CGI Shores: Regions 0-2 kb flanking CGIs, often more variable than the CGI core.
Table 1: Characteristics of Normal Tissue-Specific Methylomes
| Genomic Feature | Typical Methylation State in Somatic Cells | Functional Association | Notes on Variation |
|---|---|---|---|
| CpG Island (CGI) Promoters | Mostly unmethylated (<10%) | Active or poised gene expression; protected from methylation. | Canonical mark of active regulatory regions. |
| CGI Shores | Low to intermediate (20-60%) | Tissue-specific regulation; often inverse correlation with expression. | High variability between tissues; key tsDMR location. |
| Gene Bodies | Highly methylated (70-80%) | Transcription elongation, splice site definition, prevention of spurious initiation. | Moderate tissue-specific variation. |
| Repetitive Elements | Highly methylated (>80%) | Maintenance of genomic stability. | Hypomethylation is a hallmark of global dysregulation. |
| Enhancers | Variable, often low | Cell-type-specific activity; methylation often inversely correlates with activity. | Dynamic during differentiation. |
Disease-Associated Alterations: Patterns and Interpretation
Disease shifts the methylome from its tissue-specific baseline. Two primary alteration types are recognized:
- Global Hypomethylation: Loss of methylation in repetitive elements and gene bodies, promoting genomic instability.
- Focal Hypermethylation: Gain of methylation at specific regulatory elements, particularly CGI promoters of tumor suppressor genes (TSGs), leading to stable silencing.
Table 2: Comparative Analysis of Methylation Alterations in Disease (e.g., Cancer)
| Alteration Type | Genomic Target | Quantitative Change (vs. Normal Tissue) | Consequence | Example Genes/Regions |
|---|---|---|---|---|
| Focal CGI Hypermethylation | Promoter CGIs of TSGs | Methylation increases from <10% to >60% | Stable transcriptional silencing, loss of function. | MGMT, BRCA1, MLH1, CDKN2A |
| Global Hypomethylation | Repetitive Elements (LINE-1, Alu), Gene Bodies | Methylation decreases by 20-40% overall | Chromosomal instability, activation of transposons, oncogene activation. | LINE-1, Sat2, CAGE1 |
| Enhancer Remodeling | Tissue-specific enhancers | Gains or losses of 30-50% methylation | Altered transcriptional networks, cell identity shift. | Enhancers near MYC, SOX2 |
Pathway from Methylation Alterations to Disease Progression
Experimental Protocols for Methylome Analysis
Genome-Wide Methylation Profiling (Bisulfite Sequencing)
Principle: Sodium bisulfite converts unmethylated cytosines to uracil (read as thymine), while methylated cytosines remain unchanged. Protocol (Post-Bisulfite Conversion):
- Library Preparation: Use a bisulfite-converted DNA-compatible kit (e.g., Accel-NGS Methyl-Seq, Swift Biosciences). Include PCR duplication removal strategies.
- Sequencing: Paired-end sequencing on Illumina platform (≥30x coverage recommended).
- Bioinformatic Analysis:
- Alignment: Use bisulfite-aware aligners (Bismark, BS-Seeker2).
- Methylation Calling: Extract per-cytosine methylation ratios (methylated reads/total reads).
- DMR Identification: Use tools like DSS, methylKit, or MethCP for statistical comparison between groups, adjusting for cell type heterogeneity.
- Integration: Correlate with RNA-seq data to link promoter/enhancer DMRs with gene expression changes.
Targeted Methylation Analysis (Pyrosequencing)
Principle: Quantitative analysis of methylation at single-CpG resolution in predefined regions. Protocol:
- PCR Amplification: Design primers for bisulfite-converted DNA targeting a short region (80-150 bp). One primer is biotinylated.
- Template Preparation: Bind PCR product to streptavidin-coated beads. Denature and wash to obtain single-stranded template.
- Pyrosequencing: Load template into Pyrosequencer. Sequentially dispense nucleotides; incorporation releases light (Pyrogram). Methylation percentage at each CpG is calculated from the C/T ratio.
- Validation: Ideal for validating DMRs from genome-wide studies in large cohorts.
Workflow for Defining Tissue-Specific Methylomes
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for Methylation Analysis
| Item Name (Example) | Supplier | Function in Experiment |
|---|---|---|
| EZ DNA Methylation Kit | Zymo Research | Gold-standard bisulfite conversion, high recovery, and minimal DNA degradation. |
| Accel-NGS Methyl-Seq DNA Library Kit | Swift Biosciences | Streamlined library prep from bisulfite-converted DNA for WGBS. |
| Methylated & Non-Methylated Control DNA | MilliporeSigma / Zymo Research | Positive and negative controls for bisulfite conversion efficiency and assay specificity. |
| PyroMark PCR Kit | Qiagen | Optimized for amplification of bisulfite-converted DNA prior to pyrosequencing. |
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII) | NEB | For locus-specific or genome-wide analysis using HELP-seq or similar restriction-based approaches. |
| Anti-5mC Antibody | Diagenode / Active Motif | For methylated DNA immunoprecipitation (MeDIP) experiments. |
| DNMT/ TET Activity Assay Kits | Epigentek / Cayman Chemical | Quantify enzymatic activity of methylation writers (DNMTs) or erasers (TETs) in cell extracts. |
| Cell-Free DNA Methylation Spin Columns | Norgen Biotek | Isolation of cfDNA from plasma/serum for liquid biopsy methylation studies. |
This technical guide examines the role of CpG island (CGI) methylation in gene silencing across three distinct biological contexts: cancer, aging, and neurodevelopmental disorders. Framed within the broader thesis that CGI hypermethylation is a context-dependent regulator of transcriptional repression, this whitepaper synthesizes current findings, presents comparative quantitative data, and provides detailed experimental protocols for the field. The analysis underscores that while the molecular machinery of DNA methylation is shared, the genomic targets, functional consequences, and therapeutic implications diverge significantly among these conditions.
The prevailing thesis in epigenetic research posits that the hypermethylation of promoter-associated CpG islands is a fundamental mechanism for the heritable silencing of tumor suppressor genes in cancer. However, emerging comparative epigenomics reveals that this paradigm is context-specific. In aging, CGI methylation changes are more stochastic and tissue-specific, contributing to transcriptional noise. In neurodevelopmental disorders, dysregulation often involves hypomethylation at specific loci or defects in methylation machinery, leading to aberrant gene expression. This guide details the technical approaches to dissect these nuanced differences.
Table 1: Characteristics of CGI Methylation Across Contexts
| Feature | Cancer | Aging | Neurodevelopmental Disorders |
|---|---|---|---|
| Primary Direction | Focal hypermethylation | Global & focal hyper/hypo-methylation | Often locus-specific hypomethylation or imprinting defects |
| Genomic Target | Promoters of tumor suppressors (e.g., BRCA1, MLH1) | Polycomb-targeted promoters, heterochromatin, bivalent domains | Imprinted loci (e.g., 15q11-q13), synaptic genes, repeat elements |
| Stability | Clonally inherited, stable | Progressive, stochastic | Developmentally set, stable postnatally with possible dysregulation |
| Key Enzymes Involved | DNMT1, DNMT3A/3B overactivity | DNMT1 fidelity loss, DNMT3A/3B activity changes | DNMT3A, DNMT1 mutations; MeCP2 dysfunction |
| Functional Outcome | Uncontrolled proliferation, genomic instability | Cellular dysfunction, senescence | Altered neural connectivity, synaptic plasticity deficits |
| Potential Reversibility | High (demethylating agents) | Low to moderate | Low (critical developmental window) |
Table 2: Representative Genes and Loci with Altered CGI Methylation
| Context | Gene/Locus | Methylation Change | Associated Condition/Process |
|---|---|---|---|
| Cancer | MGMT promoter | Hypermethylation | Glioblastoma, colorectal cancer |
| Cancer | GSTP1 promoter | Hypermethylation | Prostate cancer |
| Aging | ELOVL2 promoter | Hypermethylation | Epigenetic aging clock |
| Aging | Polycomb Target Genes | Hypermethylation | Tissue aging |
| Neurodevelopmental | SNRPN (PWS/AS region) | Loss of imprinting (methylation) | Prader-Willi/Angelman Syndromes |
| Neurodevelopmental | MECP2 | Mutations in reader, not writer | Rett Syndrome |
Experimental Protocols for CGI Methylation Analysis
Bisulfite Conversion and Sequencing (Gold Standard)
Principle: Sodium bisulfite converts unmethylated cytosines to uracil, while methylated cytosines remain unchanged. Post-PCR sequencing reveals methylation status at single-base resolution.
Detailed Protocol:
- DNA Denaturation: Digest 500 ng - 2 µg genomic DNA with restriction enzymes if needed. Denature with NaOH (final 0.2-0.3 M) at 37°C for 15 min.
- Bisulfite Treatment: Add freshly prepared sodium bisulfite solution (pH 5.0) and hydroquinone. Incubate in a thermal cycler: 95°C for 30 sec, 50°C for 60 min, for 16-20 cycles. Protect from light.
- Clean-up: Use commercial bisulfite cleanup kits (e.g., Zymo Research EZ DNA Methylation kits). Desulfonate with NaOH (final 0.3 M) for 15 min at room temperature.
- Neutralization & Elution: Neutralize with ammonium acetate (pH 7.0) and precipitate with ethanol. Elute in TE buffer or nuclease-free water.
- Amplification & Sequencing: Design PCR primers specific for bisulfite-converted DNA. Amplify target CGI regions. Analyze via Sanger sequencing, pyrosequencing, or next-generation sequencing (Whole Genome Bisulfite Sequencing - WGBS).
Methylated DNA Immunoprecipitation (MeDIP)
Principle: Immunoprecipitation with an antibody against 5-methylcytosine (5-mC) to enrich methylated DNA fragments.
Detailed Protocol:
- DNA Shearing: Sonicate genomic DNA to ~100-500 bp fragments.
- Immunoprecipitation: Denature sheared DNA at 95°C for 10 min and immediately chill on ice. Incubate with anti-5-mC antibody in IP buffer (e.g., 10 mM sodium phosphate, pH 7.0, 140 mM NaCl, 0.05% Triton X-100) overnight at 4°C with rotation.
- Capture: Add pre-washed Protein A/G magnetic beads and incubate for 2 hours.
- Washing: Wash beads 3-5 times with IP buffer.
- Elution & Purification: Elute DNA with Proteinase K in elution buffer at 55°C for 2 hours. Purify DNA using phenol-chloroform extraction or columns.
- Analysis: Quantify enriched DNA via qPCR for specific loci or subject to next-generation sequencing (MeDIP-seq).
Methylation-Sensitive High-Resolution Melting (MS-HRM)
Principle: Distinguishes methylated and unmethylated DNA based on melting curve profiles post-PCR from bisulfite-converted DNA.
Detailed Protocol:
- Bisulfite Conversion: As in 3.1.
- PCR with Saturation Dye: Perform real-time PCR in the presence of a saturating DNA dye (e.g., EvaGreen) using primers flanking the CpG site of interest.
- High-Resolution Melting: After amplification, slowly increase temperature (0.1-0.2°C/sec) from 65°C to 95°C while continuously monitoring fluorescence.
- Analysis: Compare sample melting curve shapes and temperature shifts to those of standard controls (0%, 50%, 100% methylated).
Visualizations
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for CGI Methylation Research
| Item Name (Example) | Category | Function & Brief Explanation |
|---|---|---|
| EpiTect Bisulfite Kits (Qiagen) | Bisulfite Conversion | Provides optimized reagents for complete and consistent cytosine conversion with high DNA recovery. Critical for all downstream bisulfite-based assays. |
| EZ DNA Methylation Kits (Zymo Research) | Bisulfite Conversion | Similar all-inclusive kits for bisulfite conversion and clean-up, known for robustness with low-input DNA. |
| Methylated & Unmethylated Human Control DNA | Control Standards | Pre-treated DNA standards (e.g., from CpGenome) essential for calibrating assays, generating standard curves (MS-HRM, pyrosequencing), and testing antibody specificity. |
| MethylMiner Kit (Invitrogen) | MeDIP | Uses MBD-Fc protein bound to beads to capture methylated DNA, an alternative to antibody-based MeDIP. |
| Anti-5-Methylcytosine Antibody | MeDIP | Monoclonal antibody specifically recognizing 5-mC for immunoprecipitation or immunofluorescence. Key for MeDIP and dot-blot assays. |
| Methylation-Specific PCR (MSP) Primers | Assay Reagents | Custom-designed primer pairs that discriminate methylated vs. unmethylated sequences after bisulfite conversion. |
| PyroMark PCR Kits (Qiagen) | Pyrosequencing | Optimized reagents for accurate amplification and sequencing of bisulfite-converted DNA for quantitative methylation analysis. |
| SsoFast EvaGreen Supermix (Bio-Rad) | MS-HRM | A PCR mix containing a saturating dye ideal for high-resolution melting curve analysis post-amplification. |
| NEBNext Enzymatic Methyl-seq Kit | NGS Library Prep | Enables direct detection of 5-mC and 5-hmC without bisulfite conversion, reducing DNA damage and bias. |
| Methylation-Aware Aligners (e.g., Bismark, BSMAP) | Bioinformatics Software | Align bisulfite-converted sequencing reads to a reference genome and call methylation status at each CpG. |
The study of CpG island methylation as a primary mechanism of epigenetic gene silencing represents a cornerstone of modern molecular biology. Mouse models (Mus musculus) have been indispensable in elucidating the enzymes (DNMTs, TETs), regulatory complexes, and phenotypic consequences of targeted promoter hypermethylation. This whitepaper examines the profound conservation of these epigenetic mechanisms between mice and humans, highlighting the translational lessons learned while critically addressing the biological and technical limitations that can hinder direct extrapolation to human biology and therapeutic development.
Core Lessons in Epigenetic Conservation from Mouse Models
Mouse studies have robustly established foundational principles of CpG island biology.
Key Conserved Mechanisms:
- Enzyme Conservation: DNMT1 (maintenance), DNMT3A/3B (de novo), and the TET family (demethylation) are highly conserved in structure and core function.
- Sequence Specificity: Transcription factors (e.g., REST, E2F) whose binding can recruit chromatin modifiers show conserved binding sites in orthologous gene promoters.
- Silencing Logic: Hypermethylation of gene promoter CpG islands is consistently associated with a repressive chromatin state (H3K9me3, H3K27me3, loss of H3K4me3) in both species.
- X-Chromosome Inactivation: The role of Xist RNA and subsequent CpG island methylation in maintaining X-inactivation is a paradigm established in mice and confirmed in humans.
Table 1: Quantitative Conservation of Key Epigenetic Machinery
| Component | Mouse Gene | Human Ortholog | Protein Identity | Key Conserved Function | Notable Divergence |
|---|---|---|---|---|---|
| Maintenance Methyltransferase | Dnmt1 | DNMT1 | ~95% | Copies methylation patterns after DNA replication. | Isoform expression patterns in specific cell types. |
| De Novo Methyltransferase | Dnmt3a | DNMT3A | ~92% | Establishes new CpG methylation, crucial for development. | Somatic mutation hotspots in human disorders (e.g., AML) not fully recapitulated in mouse models. |
| Demethylase | Tet1 | TET1 | ~86% | Oxidizes 5mC to 5hmC, initiating active demethylation. | Expression levels and specific roles in early embryogenesis differ. |
| Methyl-CpG Binding Protein | Mecp2 | MECP2 | ~98% | Binds methylated CpGs and recruits repressive complexes. | MECP2 duplication syndrome severity is not perfectly modeled in mice. |
Experimental Protocols from Foundational Studies
Protocol 1: Bisulfite Sequencing of Target CpG Islands in Mouse vs. Human Tissues
- Objective: Compare methylation status of orthologous gene promoters.
- Sample Preparation: Isolate genomic DNA from matched tissue types (e.g., liver, cortex) from adult C57BL/6 mice and human post-mortem/biopsy samples.
- Bisulfite Conversion: Treat 500 ng DNA with sodium bisulfite (e.g., EZ DNA Methylation Kit) to convert unmethylated cytosines to uracil, while methylated cytosines remain unchanged.
- PCR Amplification: Design primers specific to the converted sequence of the CpG island of interest (e.g., Cdkn2a/p16INK4a promoter). Use high-fidelity, bisulfite-converted DNA-tolerant polymerase.
- Sequencing & Analysis: Clone PCR products, Sanger sequence multiple clones, or use next-generation sequencing. Calculate percentage methylation per CpG site. Use statistical tests (e.g., Mann-Whitney U) to compare mouse vs. human methylation profiles at each homologous site.
Protocol 2: Chromatin Immunoprecipitation (ChIP) for Repressive Marks
- Objective: Assess cross-species conservation of histone modifications at a silenced locus.
- Cross-linking & Sonication: Cross-link cells/tissue with 1% formaldehyde. Sonicate chromatin to 200-500 bp fragments.
- Immunoprecipitation: Incubate with antibodies against H3K9me3 or H3K27me3. Use species-validated antibodies. Include IgG control.
- DNA Recovery & qPCR: Reverse cross-links, purify DNA. Perform qPCR with primers spanning the methylated CpG island and a control non-silenced region (e.g., Gapdh promoter). Enrichment is calculated as % of input.
Limitations in Translating Mouse Epigenetic Findings to Humans
Despite conservation, critical limitations exist:
- Genomic Context: CpG island landscape and genomic repeat element distribution differ.
- Lifespan & Environmental Exposure: The impact of chronic, low-dose environmental exposures on the methylome over a 70+ year human lifespan is poorly modeled in short-lived mice.
- Cellular Heterogeneity: "Same tissue" comparisons are confounded by differing stromal and immune cell populations.
- Inbred vs. Outbred Genetics: Standard lab mice are inbred, reducing the genetic variability fundamental to human population studies.
- Complex Behavioral & Cognitive Phenotypes: Outcomes for neuropsychiatric disorders linked to epigenetic dysregulation are difficult to translate.
Table 2: Case Study - Translational Challenges inCdkn2a/p16INK4aSilencing
| Aspect | Mouse Model Data (C57BL/6) | Human Data | Translational Gap |
|---|---|---|---|
| Age-Dependent Methylation | Clear increase in intestinal crypts by 24 months. | Detectable in colon mucosa by 40-50 years, but more variable. | Timing and stochastic onset differ. |
| Trigger | Replicative senescence in culture; aging in vivo. | Linked to chronic inflammation (e.g., IBD), smoking. | Etiological drivers are more complex in humans. |
| Therapeutic Reversal | Demethylating agents (5-azacytidine) effectively reverse silencing in vitro. | Clinical use of DNMT inhibitors shows hypomethylation but with high toxicity and off-target effects. | Mouse models do not predict therapeutic index or systemic toxicity in humans. |
Visualizing Conserved and Divergent Pathways
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function in CpG Island Methylation Research | Key Consideration for Cross-Species Work |
|---|---|---|
| Sodium Bisulfite | Converts unmethylated cytosine to uracil for methylation status detection. | Conversion efficiency must be rigorously optimized for both mouse and human DNA, which can have differing purities and contaminants. |
| Anti-5-Methylcytosine (5mC) Antibody | Immunodetection of global or locus-specific DNA methylation (e.g., MeDIP). | Antibody specificity must be validated for both species; cross-reactivity to 5hmC can confound results. |
| DNMT Inhibitors (e.g., 5-Azacytidine) | Induces DNA hypomethylation by trapping DNMTs. | Cytotoxicity and off-target effect profiles differ markedly between mouse cell lines and primary human cells. |
| TET Enzyme Activators (e.g., Vitamin C) | Promotes active DNA demethylation by enhancing TET activity. | Dose-response and efficacy can be species- and cell type-dependent. |
| Species-Specific ChIP-Validated Antibodies | For histone marks (H3K27me3, H3K4me3) in chromatin analysis. | Antibodies validated for human samples may have lower affinity for mouse epitopes and vice-versa. |
| CRISPR/dCas9-DNMT3A/3L Fusion Systems | For targeted methylation of specific CpG islands. | gRNA design must account for sequence differences in orthologous human/mouse promoters. |
| Reduced Representation Bisulfite Sequencing (RRBS) Kit | For cost-effective, genome-wide methylation analysis at CpG-rich regions. | Enzymatic digestion (e.g., MspI) efficiency must be consistent across species' genomic DNA. |
The discovery of aberrant CpG island methylation as a pivotal mechanism for gene silencing in cancer has catalyzed the search for methylated DNA sequences as biomarkers. These biomarkers offer immense potential for early detection (diagnostic), risk stratification (prognostic), and prediction of therapy response. However, the transition from research observation to clinically useful test demands rigorous validation grounded in robust statistical and methodological frameworks. This guide details the core principles of evaluating biomarker performance, with specific application to DNA methylation biomarkers derived from gene silencing research.
Core Performance Metrics: Sensitivity, Specificity, and Predictive Values
The diagnostic accuracy of a biomarker is primarily assessed against a gold standard. For methylation biomarkers in oncology, this is often histopathological confirmation.
Table 1: Core Metrics for Binary Biomarker Tests
| Metric | Definition | Formula | Interpretation in Methylation Biomarker Context |
|---|---|---|---|
| Sensitivity (True Positive Rate) | Proportion of diseased individuals correctly identified. | TP / (TP + FN) | Ability to detect cancer when it is present (e.g., detect MGMT promoter methylation in glioblastoma). |
| Specificity (True Negative Rate) | Proportion of non-diseased individuals correctly identified. | TN / (TN + FP) | Ability to correctly identify healthy tissue or benign conditions. |
| Positive Predictive Value (PPV) | Probability that a positive test indicates true disease. | TP / (TP + FP) | Probability that a positive methylation test (e.g., SEPTIN9 in blood) truly indicates colorectal cancer. |
| Negative Predictive Value (NPV) | Probability that a negative test indicates no disease. | TN / (TN + FN) | Probability that a negative methylation test rules out disease. |
| Accuracy | Overall proportion of correct classifications. | (TP + TN) / Total | Overall test performance. |
TP: True Positive; TN: True Negative; FP: False Positive; FN: False Negative.
These metrics are intrinsically linked, as visualized in the relationship between disease prevalence, sensitivity, specificity, and predictive values.
Diagram Title: Relationship Between Prevalence, Test Metrics, and Predictive Values
The Receiver Operating Characteristic (ROC) Curve and the Area Under the Curve (AUC)
For quantitative methylation assays (e.g., methylation-specific qPCR yielding a % methylation value), a single sensitivity/specificity pair is insufficient. The ROC curve plots sensitivity (TPR) against 1-Specificity (FPR) across all possible cut-off points.
Table 2: Interpreting AUC Values for Methylation Biomarkers
| AUC Range | Diagnostic/Prognostic Utility |
|---|---|
| 0.90 – 1.00 | Excellent discrimination (e.g., a highly specific methylated marker in liquid biopsy). |
| 0.80 – 0.90 | Good discrimination. |
| 0.70 – 0.80 | Fair discrimination. |
| 0.60 – 0.70 | Poor discrimination. |
| 0.50 – 0.60 | No discrimination (test is uninformative). |
Diagram Title: ROC Curve Conceptual Graph with AUC Examples
Detailed Experimental Protocol: Validation of a Methylation Biomarker via Pyrosequencing
Objective: To quantitatively validate the methylation percentage of a specific CpG island (e.g., within the CDKN2A p16 promoter) in formalin-fixed, paraffin-embedded (FFPE) tumor samples.
Workflow Overview:
Diagram Title: Pyrosequencing Methylation Validation Workflow
Protocol Details:
- DNA Extraction & Bisulfite Conversion: Extract genomic DNA from FFPE sections using a dedicated kit (e.g., QIAamp DNA FFPE Tissue Kit). Treat 500 ng DNA with sodium bisulfite using the EZ DNA Methylation-Lightning Kit, converting unmethylated cytosines to uracil, while methylated cytosines remain as cytosine.
- PCR Amplification: Design PCR primers (one biotinylated) targeting the bisulfite-converted sequence of interest. Perform PCR in a 25 µL reaction with HotStarTaq Plus DNA Polymerase. Verify amplicon on agarose gel.
- Pyrosequencing Preparation: Bind 10 µL of biotinylated PCR product to Streptavidin Sepharose High Performance beads. Wash and denature with NaOH. Anneal the sequencing primer (0.3 µM) to the template.
- Pyrosequencing Run: Load the primer-template complex into a PyroMark Q24 cartridge containing pre-dispensed nucleotides (dATPαS, dCTP, dGTP, dTTP). Run the sequencer. The instrument sequentially dispenses nucleotides; incorporation of a nucleotide complementary to the template strand releases pyrophosphate, generating a light signal proportional to the number of nucleotides incorporated.
- Quantitative Analysis: The PyroMark Q24 software generates a pyrogram and calculates the percentage methylation at each interrogated CpG site by comparing the C/T peak heights.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Methylation Biomarker Validation
| Item | Function & Rationale |
|---|---|
| Sodium Bisulfite Conversion Kit (e.g., EZ DNA Methylation Kit) | Chemically converts unmethylated cytosine to uracil, creating sequence differences based on methylation status. Fundamental for all downstream assays. |
| Methylation-Specific PCR (MSP) Primers | Primer pairs designed to amplify either the methylated or unmethylated bisulfite-converted sequence. Used for rapid, sensitive binary detection. |
| Pyrosequencing System & Reagents | Provides quantitative, base-resolution methylation percentages across short sequences. Gold standard for validation of specific CpG sites. |
| Digital Droplet PCR (ddPCR) Probe Assays | Enables absolute quantification of rare methylated alleles in a high background of unmethylated DNA (e.g., in liquid biopsies). Offers high precision and sensitivity. |
| Next-Generation Sequencing (NGS) Panel (e.g., targeted bisulfite sequencing) | Allows for comprehensive, multiplexed analysis of methylation across many genes/regions simultaneously. Ideal for discovery and advanced validation. |
| Universal Methylated & Unmethylated Human DNA Controls | Provide essential positive and negative controls for bisulfite conversion and assay performance, ensuring accuracy and reproducibility. |
Assessing Clinical Utility: Beyond Accuracy
A biomarker with excellent sensitivity and specificity must demonstrate clinical utility—evidence that using the test improves patient outcomes or decision-making compared to standard care. For a prognostic methylation biomarker (e.g., MGMT promoter methylation predicting temozolomide response in glioblastoma), this involves:
- Clinical Validity: Demonstrating a strong, consistent association with the clinical endpoint (e.g., progression-free survival) in independent, well-designed cohorts.
- Analytical Validity: Ensuring the test itself is reliable, reproducible, and feasible in a clinical lab (CLIA/CAP certified).
- Impact on Clinical Decision-Making: Proving that test results are actionable and lead to improved net outcomes (e.g., randomized trials showing biomarker-guided therapy improves survival or quality of life).
- Health Economic Analysis: Evaluating cost-effectiveness compared to current standard pathways.
Conclusion: Validation of methylation biomarkers requires a multi-stage process from analytical confirmation to demonstration of clinical value. By adhering to rigorous standards for sensitivity, specificity, and utility assessment, researchers can translate discoveries in CpG island methylation into tools that genuinely impact patient care in diagnosis, prognosis, and personalized therapy.
1. Introduction and Thesis Context
Within the broader thesis of CpG island (CGI) hypermethylation as a key mechanism of tumor suppressor gene silencing in cancer, the emergence of DNA methyltransferase inhibitors (DNMTi) represents a paradigm-shifting therapeutic strategy. Unlike cytotoxic agents, DNMTi like azacitidine and decitabine aim for epigenetic reprogramming, requiring distinct biomarkers to assess their biological and clinical efficacy. This whitepaper posits that dynamic, therapy-induced changes in CGI methylation at specific loci serve as superior pharmacodynamic biomarkers, correlating more directly with target engagement and transcriptional reactivation than traditional clinical metrics alone. Their precise assessment is critical for optimizing dose schedules, identifying responsive patient populations, and developing next-generation epigenetic therapies.
2. Core Biomarker Loci and Quantitative Data Summary
Research has identified recurrently hypermethylated CGIs that undergo demethylation upon effective DNMTi treatment. The degree of change is quantifiable and correlates with outcomes. The following table summarizes key biomarker loci and associated data from recent clinical and preclinical studies.
Table 1: Key CGI Methylation Biomarkers for DNMTi Response Assessment
| Gene Locus (CGI) | Biological Function | Baseline Methylation in Disease | Post-DNMTi Methylation Change (Representative) | Correlated Outcome |
|---|---|---|---|---|
| CDKN2A (p14/ARF, p16) | Cell cycle regulation | 60-90% in MDS, AML | -20% to -50% (after 1 cycle) | OS, hematologic improvement |
| MLH1 | DNA mismatch repair | 10-50% in colorectal, endometrial cancers | -15% to -40% | Gene re-expression, restored repair |
| SFRP1 | WNT signaling inhibitor | 70-100% in solid tumors | -25% to -60% | Reduced proliferation in vitro |
| LINE-1 | Repetitive element (surrogate) | Variable hypomethylation globally | +5% to +15% (genomic hypomethylation reversal) | Non-specific pharmacodynamic marker |
| HOXA9 | Developmental transcription factor | >80% in AML | -30% to -70% | Clinical response in AML |
3. Detailed Experimental Protocols for Biomarker Assessment
Protocol 3.1: Pyrosequencing for Quantitative Methylation Analysis (Post-Bisulfite Conversion) Objective: To obtain quantitative, base-resolution methylation percentages for specific CpG sites within a target CGI. Materials: DNA sample (pre- and post-treatment), EZ DNA Methylation-Lightning Kit (Zymo Research), PCR primers (bisulfite-converted specific), PyroMark PCR Kit (Qiagen), PyroMark Q96 MD system. Procedure:
- Bisulfite Conversion: Treat 500 ng genomic DNA using the Lightning Kit per manufacturer’s protocol, converting unmethylated cytosines to uracil.
- PCR Amplification: Design primers flanking the target CGI region (avoiding CpG sites). Amplify 20 ng converted DNA with biotinylated reverse primer.
- Pyrosequencing Preparation: Bind biotinylated PCR product to Streptavidin Sepharose HP beads, denature with NaOH, and wash.
- Sequencing: Anneal sequencing primer to the single-stranded template. Load into the Pyrosequencer with dispensation order of nucleotides. The light signal generated upon nucleotide incorporation is proportional to the number of C/T at each CpG, yielding a precise % methylation.
Protocol 3.2: Next-Generation Sequencing (NGS)-Based Targeted Bisulfite Sequencing Objective: For high-throughput, multiplexed quantification of methylation across multiple CGI loci in many samples. Materials: Bisulfite-converted DNA, SureSelectXT Methyl-Seq Target Enrichment System (Agilent) or similar amplicon-based NGS panel, Illumina sequencing platform. Procedure:
- Library Preparation & Target Enrichment: Following bisulfite conversion, prepare sequencing libraries from fragmented DNA. Hybridize libraries to biotinylated RNA probes designed for target CGIs.
- Capture and Amplification: Capture probe-bound fragments, wash, and perform PCR amplification.
- Sequencing & Analysis: Sequence on an Illumina platform (150bp paired-end). Align reads to a bisulfite-converted reference genome using tools like Bismark or BSMAP. Calculate methylation percentage as (C reads / (C reads + T reads)) at each CpG site.
4. Signaling Pathways and Mechanistic Workflow
Diagram 1: DNMTi Action and Biomarker Genesis Pathway
Diagram 2: Biomarker Assessment Workflow
5. The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for CGI Methylation Biomarker Studies
| Reagent/Kit | Vendor Examples | Primary Function in Workflow |
|---|---|---|
| DNA Bisulfite Conversion Kit | EZ DNA Methylation-Lightning Kit (Zymo), EpiTect Fast (Qiagen) | Converts unmethylated cytosine to uracil for sequence discrimination, critical for all downstream assays. |
| Pyrosequencing System & Kits | PyroMark Q96 MD, PyroMark PCR Kit (Qiagen) | Enables quantitative, base-resolution methylation analysis at specific CpG sites post-PCR. |
| Targeted Methyl-Seq Panels | SureSelect Methyl-Seq (Agilent), Twist NGS Methylation System | Allows multiplexed, deep sequencing of custom or pre-designed panels of CGI regions. |
| Methylation-Specific qPCR Assays | TaqMan Methylation Assays (Thermo Fisher) | For rapid, quantitative screening of methylation status at a single, predefined locus. |
| Universal Methylated & Unmethylated DNA Controls | EpiTect PCR Control DNA (Qiagen) | Essential positive/negative controls for bisulfite conversion efficiency and assay validation. |
| Next-Gen Sequencing Library Prep for FFPE | Accel-NGS Methyl-Seq DNA Library Kit (Swift Biosciences) | Optimized for degraded DNA from formalin-fixed, paraffin-embedded (FFPE) clinical archives. |
| DNMT Inhibitors (for in vitro validation) | Decitabine, Azacitidine (Sigma, Selleckchem) | Used in cell line models to establish baseline biomarker dynamics and dose-response relationships. |
This whitepaper provides a technical framework for the independent validation of CpG island methylation findings using publicly available genomic and epigenomic datasets. Within the broader thesis context of CpG island methylation and its role in gene silencing, the integration of data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) is presented as a critical, cost-effective step for confirming experimental results and ensuring robust, reproducible science. This guide details methodologies, workflows, and best practices tailored for researchers and drug development professionals.
Research into CpG island methylation, particularly its hypermethylation in promoter regions leading to transcriptional silencing of tumor suppressor genes, is a cornerstone of cancer epigenetics. Initial discoveries often arise from focused, in-house experiments. However, the transition from a novel finding to a biologically and clinically validated insight requires confirmation across independent cohorts. Public repositories like TCGA (The Cancer Genome Atlas) and GEO (Gene Expression Omnibus) offer vast, multi-omics datasets from thousands of patients and diverse experimental conditions. Leveraging these resources allows for hypothesis testing, assessment of prevalence across cancer types, correlation with clinical outcomes, and integration with complementary data layers (e.g., gene expression, copy number variation).
Dataset Landscape: TCGA and GEO
The Cancer Genome Atlas (TCGA)
TCGA is a landmark project profiling genomic, epigenomic, transcriptomic, and proteomic data from over 20,000 primary cancer and matched normal samples across 33 cancer types. For methylation research, its most critical component is the Illumina Infinium HumanMethylation450K and MethylationEPIC (850K) array data, providing quantitative methylation beta-values for hundreds of thousands of CpG sites.
Gene Expression Omnibus (GEO)
GEO is a public functional genomics data repository supporting array- and sequence-based data. It is an invaluable source for independent validation as it hosts thousands of user-submitted methylation datasets (from arrays and bisulfite sequencing) alongside gene expression profiles from diverse studies, organisms, and disease states.
Table 1: Core Features of TCGA and GEO for Methylation Validation
| Feature | The Cancer Genome Atlas (TCGA) | Gene Expression Omnibus (GEO) |
|---|---|---|
| Primary Data Type | Curated, multi-omics projects | User-submitted, heterogeneous studies |
| Methylation Platform | Primarily Illumina 450K/850K arrays | Diverse: Illumina arrays, RRBS, WGBS, etc. |
| Sample Number | Large cohorts per cancer type (100-1000s) | Varies widely per series (10s-100s) |
| Clinical Data | Standardized, extensive clinical annotations | Often limited, dependent on submitter |
| Access Method | Programmatic (e.g., TCGAbiolinks, GDCRNATools) or portals (cBioPortal, UCSC Xena) | Web interface or programmatic (GEOquery, SRAtoolkit) |
| Best For | Assessing prevalence, clinical correlations, and pan-cancer analysis within a standardized framework | Validating findings in specific disease models, treatments, or non-cancer contexts. |
Technical Workflow for Data Integration and Validation
The following protocol outlines a standard workflow for validating a candidate hypermethylated CpG island/gene identified from a primary experiment.
Hypothesis Definition & Target Identification
- Input: A list of candidate genes showing promoter CpG island hypermethylation and associated gene silencing in your primary model (e.g., cell line or patient cohort).
- Goal: Confirm that methylation of these targets is recurrent, specific to the disease state, and inversely correlated with gene expression in independent public data.
Data Acquisition & Preprocessing
Protocol A: Accessing and Processing TCGA Methylation Data
- Tool Selection: Use the
TCGAbiolinksR/Bioconductor package. - Query Data: Use
GDCquery()to search for the desired project (e.g., "TCGA-BRCA") and data type ("DNA methylation"). - Download: Execute
GDCdownload()followed byGDCprepare()to load data into R as aSummarizedExperimentobject. - Preprocessing: Perform normalization (e.g., functional normalization via
preprocessFunnorminminfipackage) and filter probes: remove those cross-reactive, on sex chromosomes, or with detection p-value > 0.01. - Annotation: Map CpG probe IDs (e.g., cg00035864) to genomic coordinates and nearby genes using platforms like IlluminaHumanMethylation450kanno.ilmn12.hg19.
Protocol B: Accessing and Processing GEO Methylation Data
- Identification: Search GEO using keywords (e.g., "methylation breast cancer GSE12345").
- Metadata Review: Critically assess the
GEO Series (GSE)metadata for platform, sample groups, and experimental design. - Download: Use
GEOquery::getGEO()to download the series matrix and platform annotation files directly into R. - Normalization: Apply appropriate normalization (e.g., BMIQ for 450K arrays using
wateRmelonpackage) consistent with the original study's methods.
Integrative Bioinformatic Analysis
Key Experiment 1: Differential Methylation Analysis
- Methodology: For a case-control design (e.g., tumor vs. normal), use linear models. In R, employ
limmafor array-based data.- Create a design matrix:
design <- model.matrix(~0 + sample_type + other_covariates). - Fit the model:
fit <- lmFit(methylation_beta_values, design). - Specify contrasts:
cont.matrix <- makeContrasts(Tumor_vs_Normal = Tumor - Normal, levels=design). - Perform empirical Bayes moderation:
fit2 <- contrasts.fit(fit, cont.matrix); fit2 <- eBayes(fit2). - Extract results:
topTable(fit2, coef="Tumor_vs_Normal", number=Inf, p.value=0.05).
- Create a design matrix:
- Validation Metric: Confirm your candidate probes/genes are significantly hypermethylated (adjusted p-value < 0.05, delta beta > 0.2).
Key Experiment 2: Methylation-Expression Correlation
- Methodology: Integrate matched methylation and RNA-seq (or microarray) expression data.
- Data Alignment: Ensure sample pairing using TCGA barcodes. For GEO, use sample identifiers from the GSE matrix.
- Extraction: For your candidate gene, extract the beta-values of promoter-associated CpG probes and the corresponding gene's normalized expression values (e.g., FPKM from RNA-seq).
- Statistical Test: Calculate Pearson or Spearman correlation coefficients for each probe-gene pair.
- Visualization: Generate scatter plots with a trend line.
- Validation Metric: A significant negative correlation (p-value < 0.05, r < -0.3) supports the functional relationship between hypermethylation and gene silencing.
Table 2: Example Validation Results for a Fictional Tumor Suppressor Gene "TSG1"
| Data Source | Cancer Type | Probe ID | Avg. Methylation (Tumor) | Avg. Methylation (Normal) | Delta Beta | Adj. P-value | Correlation with Expression (r) | Correlation P-value |
|---|---|---|---|---|---|---|---|---|
| TCGA-COAD | Colon Adenocarcinoma | cg12345678 | 0.72 | 0.18 | 0.54 | 1.2e-15 | -0.65 | 3.5e-10 |
| GSE12345 | Colorectal Cancer | cg12345678 | 0.68 | 0.22 | 0.46 | 5.8e-08 | -0.58 | 2.1e-05 |
| TCGA-BRCA | Breast Cancer | cg12345678 | 0.41 | 0.25 | 0.16 | 0.03 | -0.22 | 0.12 |
Visualizing the Validation Workflow and Molecular Relationships
Title: Public Data Validation Workflow
Title: CpG Methylation Leads to Gene Silencing
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for Methylation Validation Studies
| Item / Solution | Function / Purpose in Validation Pipeline |
|---|---|
| R/Bioconductor Packages (TCGAbiolinks, GEOquery, minfi, limma) | Core software tools for programmatic data download, preprocessing, normalization, and differential analysis of public methylation data. |
| Illumina Infinium Methylation BeadChip Arrays (450K/EPIC) | The dominant platform generating data in TCGA and many GEO series. Understanding their probe design and biases is essential for analysis. |
| Bisulfite Conversion Reagents (e.g., EZ DNA Methylation Kits) | The gold-standard chemical treatment that converts unmethylated cytosines to uracil, allowing methylation status to be read as sequence differences. Critical for validating key findings in the lab. |
| Pyrosequencing Assay & Primers | A quantitative, high-resolution method for validating the methylation levels at specific CpG sites identified from public data analysis in independent patient samples. |
| Methylation-Specific PCR (MSP) Primers | A rapid, sensitive method for detecting the presence of hypermethylated alleles at a specific locus, useful for clinical sample screening. |
| UCSC Genome Browser / IGV | Visualization tools to map CpG probe locations, view CpG island annotations, and integrate public data tracks with your own findings. |
| cBioPortal / UCSC Xena | User-friendly web portals for quick, interactive exploration of TCGA data, including methylation and clinical correlations, without programming. |
Conclusion
CpG island methylation stands as a cornerstone of epigenetic gene regulation, with its disruption underpinning a vast array of human diseases, most notably cancer. This review has synthesized knowledge from foundational biology through to advanced clinical applications, emphasizing that robust methodological execution and rigorous validation are paramount for meaningful discovery. The future of this field lies in further elucidating the upstream triggers of aberrant methylation, refining single-cell and liquid biopsy technologies for early detection, and developing next-generation, targeted epigenetic therapies that can reverse pathogenic silencing with greater specificity. For researchers and drug developers, a deep understanding of CGI methylation dynamics offers a powerful lens through which to diagnose, understand, and ultimately treat complex epigenetic diseases.